
You have full access to this article via your institution.
In a recent paper published in Nature, Tan et al. identify phosphatidylinositol-3,5-bisphosphate (PtdIns(3,5)P₂) as an endogenous ligand that licenses cGAMP-induced STING activation. The study offers a mechanistic explanation on how full-scale STING signaling activation is coupled to its oligomerization on the post-Golgi compartment.
Innate immune defense relies on surveillance mechanisms that detect microbial invasion and cellular injury. In vertebrates, the cGAS-STING pathway senses and responds to double-stranded DNA (dsDNA) aberrantly present in the cytosol. Upon binding dsDNA, cyclic GMP-AMP synthase (cGAS) produces the cyclic dinucleotide second messenger cyclic GMP-AMP (cGAMP), which engages the stimulator of interferon genes (STING) to trigger downstream signaling.1 STING homologues are broadly conserved across metazoans and can be activated by cyclic dinucleotides produced by diverse cGAS-like receptors that sense nucleic acids, primarily dsDNA, but in some cases RNA, and possibly other ligands.
STING is a transmembrane protein that resides in the endoplasmic reticulum (ER) as a homodimer under the resting state. cGAMP binding induces a pronounced conformational rearrangement that promotes further oligomerization of STING dimers. Oligomerized STING exits the ER and traffics through the Golgi apparatus to the trans-Golgi network (TGN) and post-Golgi vesicles before ultimately being degraded in lysosomes. This membrane trafficking itinerary is tightly coupled to downstream signaling events of the cGAS-STING pathway. Upon arrival at the TGN, higher-order STING oligomers recruit TANK-binding kinase 1 (TBK1), enabling TBK1-dependent phosphorylation of STING and IRF3 and thereby driving type I interferon production.2 In parallel, TBK1 activates NF-κB signaling, leading to the expression of proinflammatory cytokines. cGAMP binding also opens a proton channel within the STING dimer, resulting in proton leakage from the acidic post-Golgi compartment, which activates V-ATPase-dependent non-canonical autophagy and lysosomal biogenesis.
A longstanding question is why STING-mediated signaling strictly requires the trafficking of STING oligomers from the ER to the Golgi. One explanation is the presence of compartment-specific licensing factors that promote higher-order STING oligomerization at the TGN and post-Golgi vesicles. Supporting this model, the aberrant accumulation of STING in the Golgi apparatus due to defects in COPI coatomer complex renders STING hypersensitive to cGAMP stimulation.3 Several mechanisms have been proposed to underlie the compartment-specific licensing model. For instance, STING is palmitoylated at two membrane-proximal cysteine residues in the Golgi apparatus, which enhances its oligomerization and signaling.4 In addition, PtdIns4P, which is highly enriched in the Golgi compartment, has been proposed to directly or indirectly promote STING signaling.5,6 However, direct biochemical and structural evidence for a defined endogenous “licensing” factor has been lacking.
To address this gap, Tan and colleagues7 mapped the STING interactome and unexpectedly found all three components of the PIKFYVE complex,8 namely PIKFYVE, VAC14 and FIG4, constitutively associated with STING. PIKFYVE is the sole kinase that synthesizes PtdIns(3,5)P₂ from PtdIns3P. To assess whether PtdIns(3,5)P₂ influences STING signaling, the authors disrupted PIKFYVE genetically and pharmacologically. Because PtdIns(3,5)P₂ is essential for cell viability, CRISPR targeting of PIKFYVE yielded hypomorphic clones that retained low residual PIKFYVE activity, which was sufficient to support cGAMP-induced STING activation. Further depletion by siRNA or pharmacological inhibition of PIKFYVE abolished cGAMP‑induced phosphorylation of STING and TBK1. Reintroducing wild‑type PIKFYVE restored signaling, whereas a kinase‑dead mutant did not. These findings indicate that PtdIns(3,5)P₂ is required for STING activation in cells.
The team further reconstituted STING signaling in vitro using ER‑derived liposomes containing STING and TBK1 and supplemented with various phosphoinositides. Among these, PtdIns(3,5)P₂ most strongly potentiated cGAMP‑induced phosphorylation of STING and TBK1. To test whether STING binds PtdIns(3,5)P₂ directly, they measured fluorescence resonance energy transfer (FRET) between EGFP-tagged full‑length STING and fluorescently labeled PtdIns(3,5)P₂, and observed a specific dose‑dependent FRET signal. Together, these experiments established PtdIns(3,5)P₂ as a ligand of STING.
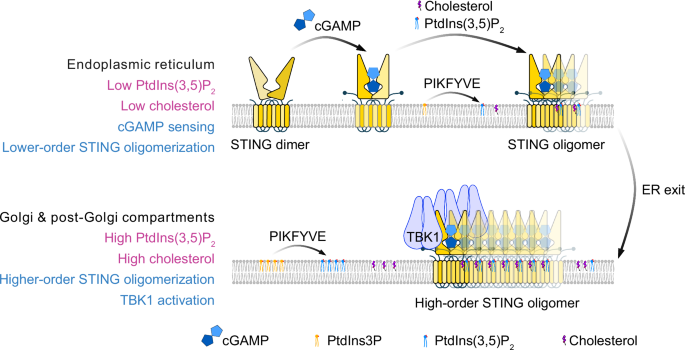
In a companion study, Li et al.9 obtained a cryo‑electron microscopy structure of full‑length STING in complex with PtdIns(3,5)P₂ and cholesterol. They located a positively charged groove between two adjacent STING dimers where the phosphoinositide headgroup binds; cholesterol sits nearby and together they act as a “molecular glue” that stabilizes side‑by‑side packing of STING dimers, thereby facilitating STING oligomerization (Fig. 1). Mutations that neutralize this pocket, such as K20A/R71A, abolished PtdIns(3,5)P₂ binding and severely impaired STING trafficking, TBK1 activation and interferon induction. Interestingly, the structural study suggests that PtdIns(4,5)P2 can also bind STING and promote STING activation in vitro. However, the constitutive association between STING and the PIKFYVE complex supports a more specific role for PtdIns(3,5)P2 in cells, at least before STING oligomers gain access to other phosphoinositides during trafficking.

At the ER, STING dimers bind cGAMP and form lower-order oligomers with limited PtdIns(3,5)P2 and cholesterol. A small pool of the lipid kinase PIKFYVE constitutively associates with STING and supports local PtdIns(3,5)P2 synthesis, which is required for STING exit from the ER. Following trafficking to the Golgi and post-Golgi compartments, higher levels of PtdIns(3,5)P2 and cholesterol further stabilize interdimer interfaces, promoting higher-order STING oligomerization and robust TBK1 activation.
Finally, Tan et al. examined the spatial and temporal requirements for PtdIns(3,5)P₂ during signaling. Complete loss of PIKFYVE trapped STING in the ER, whereas partial inhibition allowed Golgi trafficking but prevented TBK1 phosphorylation. In vitro, PtdIns(3,5)P₂ was dispensable for TBK1 recruitment to STING but was essential for TBK1 autophosphorylation. These findings indicate that low levels of PtdIns(3,5)P₂ at the ER are sufficient for STING to exit, whereas higher concentrations at post‑Golgi compartments are needed for full activation (Fig. 1).
In summary, Tan et al. together with the companion study from Li et al. identify PtdIns(3,5)P2 as an endogenous ligand of STING that licenses STING signaling by cGAMP. Their work provides a mechanistic framework explaining how STING activation is spatially coupled to membrane trafficking. The unique dependence of STING signaling on PtdIns(3,5)P2 in cells is intriguing as other phosphoinositides are also enriched in Golgi and post-Golgi compartments, and the positively charged juxtamembrane region of STING does not seem to be very selective. Under certain conditions, other phospholipids (e.g. PtdIns(4,5)P2 and PtdIns4P) may also facilitate STING oligomerization.10 Whether the specific interaction between STING and PtdIns(3,5)P2, or with the PIKFYVE complex, confers a unique advantage in innate immune defense, and whether similar principles apply to other metazoan STING homologues, remains to be determined.