
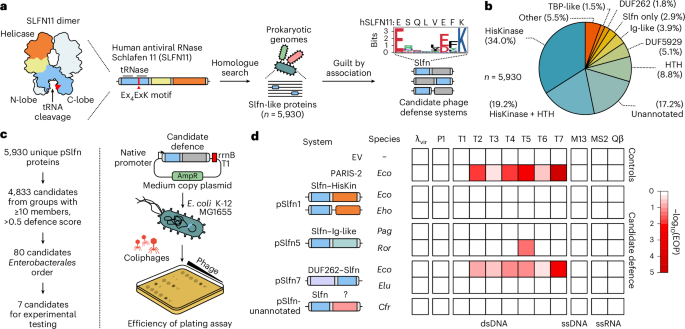
Bacterial Schlafen proteins mediate phage defence
The human SLFN11 protein has an N-terminal ribonuclease (RNase) domain, a linker domain and a C-terminal DNA/RNA helicase domain4. The N-terminal domain consists of two lobes (N- and C-lobe), where the C-lobe contains the ribonuclease active site with a catalytic Ex4ExK motif that is essential for tRNA cleavage4 (Fig. 1a). To identify prokaryotic Slfn homologues, we searched for Slfn nuclease domains in reference prokaryotic genomes and identified 5,930 unique (9,937 total) protein sequences across 4 archaeal and 34 bacterial phyla (Fig. 1a, Extended Data Fig. 1a and Supplementary Table 1). Subsequent multiple sequence alignments show a highly conserved Ex4(E/D)xK sequence motif in the predicted active site, which matches the catalytic residues of SLFN11, suggesting a conserved enzymatic mechanism (Fig. 1a and Extended Data Fig. 1b).

a, The computational approach to identify prokaryotic homologues of Schlafen nucleases (Slfn) with predicted roles in phage defence. This search found 5,930 unique Slfn protein sequences (100% CD-HIT cut off). The Weblogo plot shows the conserved Ex4(E/D)xK motif in prokaryotic Slfn domains. b, Annotation of domains fused to Slfn in the 5,930 identified proteins. c, Experimental approach to test for immune activities of Slfn proteins. d, Immune activities of tested Slfn proteins (top to bottom) against a panel of phages that infect E. coli (left to right). Cfr, Citrobacter freundii; Eco, Escherichia coli; Eho, Enterobacter hormaechei; Elu, Enterobacter ludwigii; Pag, Pantoea agglomerans; Ror, Raoultella ornithinolytica; dsDNA, double-stranded DNA; ssDNA, single-stranded DNA; ssRNA, single-stranded RNA; λvir, virulent mutant of phage λ.
Source data
Prokaryotic defence genes frequently cluster in the host genome, forming genetic neighbourhoods known as defence islands15,19. To determine whether pSlfn proteins reside in defence islands, we performed a genetic neighbourhood analysis and found that 69.6% of identified pSlfn genes are located within a ±10-kb distance to at least one known defence gene (Extended Data Fig. 1a and Supplementary Table 1). On average, identified pSlfn genes co-localize with 1.9 defence genes, which is comparable to AriB (2.74), the nuclease effector of the phage anti-restriction-induced system (PARIS)20,21, and approximately tenfold higher than the housekeeping genes rpL3 (0.13) and L1p (0.19) (Extended Data Fig. 1c). This genetic association suggested that pSlfns may function in phage defence. Notably, 30.4% of identified pSlfn genes were not associated with defence genes, suggesting that in some instances, pSlfn domains may have non-immune functions or that these pSlfn genes are in defence hot spots that have not been annotated yet21,22.
Phage defence systems often function through sensors that detect infection and effectors that execute the immune response. These functional modules can be encoded within a single gene or split across multiple genes23,24. We found that 97.1% of identified pSlfn domains are fused to other protein domains, where pSlfns most probably act as effectors, while the identified fused domains may function as sensors (Fig. 1b). To classify the diversity of domain architectures among pSlfn proteins, we used a combination of sequence and structural homology-based computational methods, identifying 55 distinct domain compositions, which we designated pSlfn1 through pSlfn55 (Supplementary Table 2). The most frequent domain compositions include fusions of pSlfn to histidine kinase (HisKinase, 34.0%), HisKinase and helix-turn-helix domains (HisKinase–HTH, 19.2%), HTH domains (8.8%), DUF5929 (5.1%), domains with immunoglobulin-like fold (Ig-like, 3.9%), DUF262 (1.8%) and TBP-like domains (1.5%) (Fig. 1b). Of 5,930 identified pSlfns, 1,020 (17.2%) are fused to protein domains that show no substantial homology to the domains annotated in the Pfam and Evolutionary Classification of protein Domains (ECOD) databases25,26. We further grouped these proteins into 558 sequence similarity clusters (Supplementary Table 2). This diversity of domain compositions in pSlfn proteins probably represents combinations of the core Slfn effector module with various sensors that respond to different cues and regulate its nuclease activity.
To test whether pSlfn genes confer phage defence, we focused on four domain architectures found in Enterobacterales, comprising fusions with HisKinase, Ig-like, DUF262 and unannotated domains. We manually picked two systems from each group, prioritizing those with the most neighbouring defence genes and greatest amino acid sequence divergence (Extended Data Fig. 1d and Supplementary Table 3). One construct failed during synthesis, leaving seven systems. We expressed these in the E. coli K-12 strain MG1655 under their native promoters and challenged the cells with a panel of diverse E. coli phages (Fig. 1c,d and Supplementary Table 4). As a positive control, we used a plasmid encoding the E. coli B185 PARIS-2 immune system21, while an empty vector (EV) served as a negative control. In the tested subset, two pSlfn genes provided defence against T phages (Fig. 1d and Extended Data Fig. 1e,f). We found that the DUF262–Slfn fusion from E. coli UC4224 (EcoSlfn7) significantly reduced the efficiency of plating (EOP) of a broad range of phages, with the most robust activity against the T7 phage (538.5 ± 194.4-fold EOP reduction, P = 6.9 × 10−4). By contrast, the Slfn fusion with Ig-like domain from Raoultella ornithinolytica (RorSlfn5) protected E. coli only against T5 phage, reducing the EOP by 22.3 ± 1.0-fold (P = 3.0 × 10−11) (Extended Data Fig. 1f). Overall, these data demonstrate that prokaryotic Schlafen proteins protect bacteria from phage infection and exhibit distinct phage specificities that are probably determined by their fused domains.
RorSlfn5 reduces the productivity of the T5 phage
To elucidate the mechanisms that underpin the immune function of pSlfn proteins, we focused on RorSlfn5 defence, which features an uncharacterized Ig-like domain, suggesting a mode of phage sensing that was not previously described. We challenged MG1655 cells with T5 phage at different multiplicities of infection (MOIs) and monitored cell growth over time. At low MOIs (0.01 and 0.1), cells expressing RorSlfn5 survived the infection compared with cells without the defence system, whereas infections at high MOIs (5 and 10) resulted in all cultures collapsing approximately 60 min post infection, which agrees with the reported lysis time for T5 phage27 (Fig. 2a,b). Further, at a MOI of 10, the phage titre in supernatants of RorSlfn5-expressing cells at 60 min post infection was reduced by 59.3 ± 8.6% compared with the no defence control (P = 7.6 × 10−3; Extended Data Fig. 2a). Alanine substitutions in the catalytic Ex₄DxK motif (E15A, D20A) of the Slfn domain abolished defence at low MOIs and rescued phage titre at a high MOI, indicating that the nuclease activity of RorSlfn5 is required for the phage defence (Fig. 2c and Extended Data Fig. 2a).

a–c, The growth kinetics of E. coli K-12 MG1655 cells without the defence (a), with RorSlfn5 (b) or the nuclease-dead RorSlfn5E15A, D20A mutant (c) following T5 bacteriophage infection at various MOIs. Each line shows a biological replicate (n = 4). d,e, The ECOI (d) and average burst size (e) of T5 phage in RorSlfn5-expressing E. coli MG1655 cells compared with the EV control. The data are shown as the mean of three biological replicates ± s.d. A two-sided Welch’s t-test was used to compare the experimental and control groups. f, An experimental approach to identify viral triggers of RorSlfn5. g, Top: plaque assays with RorSlfn5-escaping T5 phages (esc1–esc4). Bottom: mapping of mutations in T5 phage escapers compared with a reference phage genome. The vertical lines indicate mutations found in the laboratory T5 phage stock compared with the reference NCBI sequence. Vertical orange box highlights mutations present in escaper phages but not in the original viral stock. h, A toxicity assay in MG1655 cells co-transformed with the RorSlfn5 plasmid and a plasmid for arabinose-inducible expression of T5.142 protein. Tenfold serial dilutions were spotted on agar plates with 0.2% glucose (−) or 0.2% arabinose (ara) (+). pBAD-GFP was used as a control for arabinose-dependent induction.
Source data
The collapse of RorSlfn5-expressing cultures at high MOIs and reduced phage titres suggested that the phage defence may act by triggering abortive infection, killing the host cell before phage replication is completed28,29. To test this hypothesis, we performed the efficiency of centre of infection (ECOI) assay, which quantifies the number of productive infections that release at least one infectious progeny phage30. However, we found no significant difference in the number of infective centres between RorSlfn5-expressing cells and the EV control, indicating that RorSlfn5 does not abort T5 phage infection (P = 0.94; Fig. 2d). Reduced EOP (Fig. 1d) and lower phage titres with no changes in ECOI suggest that RorSlfn5 reduces T5 phage productivity without completely eliminating the phage31. Consistent with this, the average burst size of T5 was reduced by 81.2 ± 3.0% in RorSlfn5-expressing cells compared with the control (P = 3.5 × 10−4; Fig. 2e). Taken together, these assays indicate that RorSlfn5 restricts T5 phage through a non-abortive mechanism that acts downstream of infection, limiting infectious phage progeny.
T5 tail assembly chaperone triggers RorSlfn5 phage defence
To determine the viral trigger for the RorSlfn5 defence, we isolated T5 phages that escaped the immunity and sequenced their genomes (Fig. 2f). Four viral clones that we sequenced contained three distinct missense mutations (that is, D18G, D68G and I72T) in the T5.142 gene, which encodes for the phage tail assembly chaperone (NCBI accession: YP_006970.1)32 (Fig. 2g and Supplementary Table 5). To test whether the T5.142 protein triggers RorSlfn5 defence, we cloned the viral gene and its escape variants into high-copy plasmid vectors under the control of the arabinose-inducible promoter (pBAD; Fig. 2h). Expression of T5.142 in cells co-transformed with the RorSlfn5 plasmid resulted in toxicity, demonstrating that this viral protein is sufficient to trigger the defence. By contrast, cells expressing a catalytically inactive mutant of RorSlfn5 (RorSlfn5E15A, D20A) showed no growth defect compared with uninduced cells, indicating that the toxicity is mediated by the T5.142-triggered nuclease activity of RorSlfn5. In addition, escape mutations in the viral T5.142 protein rescued toxicity. Notably, while the T5.142D18G variant reduced toxicity, it did not completely abolish it, suggesting that, in our experimental set up, it is less effective at evading RorSlfn5 defence than D68G and I72T mutations.
The immunoglobulin-like domain of pSlfn5 is a phage sensor
Homologues of pSlfn5 from Pantoea agglomerans (PagSlfn5; 39.6% sequence identity to RorSlfn5) and Serratia fonticola (SfoSlfn5; 79.6% sequence identity to RorSlfn5) did not protect E. coli from T5 phage or any other phage tested (Extended Data Figs. 1f and 2b). The sequence divergence between these proteins is primarily driven by their C-terminal domains, while the N-terminal Slfn nuclease domains are more conserved (Extended Data Fig. 3a). The AlphaFold-predicted structure of RorSlfn5 suggests that its C-terminal domain has an Ig-like β-sandwich fold with structural similarity to human integrin ectodomains (Extended Data Fig. 3b,c and Supplementary Table 6). These observations led us to hypothesize that the Ig-like domains of pSlfn5 proteins function as sensor modules that recognize viral proteins, and thereby PagSlfn5 and SfoSlfn5 might recognize T5.142 homologues from other phages.
To test this, we synthesized genes encoding six representative homologues of the T5 phage tail assembly chaperone (Fig. 3a). In addition to T5.142, RorSlfn5 was triggered by expression of homologues from Salmonella phage S114, Pantoea phage vB_PagS_AAS21 and an unclassified phage from a human gut metagenome (Fig. 3b and Extended Data Fig. 2c). Consistent with the lack of defence against the T5 phage, PagSlfn5 and SfoSlfn5 were not activated by expression of T5.142, while the tail assembly chaperone from the human gut metagenome triggered all tested pSlfn5 homologues (Fig. 3b and Extended Data Fig. 2d,e). None of the identified triggers caused toxicity when expressed alone or co-expressed with a nuclease-dead RorSlfn5E15A, D20A mutant, confirming that toxicity is mediated by the nuclease activity of pSlfn5 (Extended Data Fig. 2f,g). Together, these findings demonstrate that pSlfn5 proteins recognize tail assembly chaperones from phages in the Demerecviridae family, with different pSlfn5 homologues exhibiting distinct trigger specificities.

a, A maximum-likelihood phylogenetic tree of T5.142 homologues. Selected representatives are listed for each homologue cluster and indicated with black dots. b, A summary of toxicity assays in MG1655 cells co-transformed with pSlfn5 (top to bottom) and T5.142 homologues from various phages (left to right) shown in Extended Data Fig. 2. The experiment was performed in three biological replicates and the results for one representative replicate are shown. The numbers correspond to the homologue clusters in a. c,d, Toxicity (c) and EOP assay (d) with chimeric pSlfn5 proteins with swapped Ig-like domains. The toxicity assays (Extended Data Fig. 4) were repeated independently three times with similar results. The data are shown for one replicate. EOP assay results are shown as mean ± s.d. of three biological replicates. The white dots show the average of three technical replicates for each biological replicate. A one-way ANOVA with post-hoc Tukey HSD test was used to compare the experimental groups. Sfo, Serratia fonticola.
Source data
To confirm the sensor function of Ig-like domains in pSlfn5 defence, we created chimeric proteins by swapping the Ig-like domains of RorSlfn5 and SfoSlfn5. Cellular toxicity assays showed that phage trigger specificity was transferred along with the sensor domain (Fig. 3c and Extended Data Fig. 4). Furthermore, the chimeric protein containing the nuclease domain of SfoSlfn5 and the RorSlfn5 sensor (Sfo–Ror chimera) gained protection against T5 phage, whereas changing the Ig-like domain of RorSlfn5 for that of SfoSlfn5 (Ror–Sfo chimera) abolished defence against T5 (Fig. 3d). Wild-type (WT) RorSlfn5 reduced T5 phage EOP by 18.9 ± 2.1-fold (P = 3.5 × 10−9), while the Sfo–Ror chimera was ~3.6-fold less efficient and reduced EOP by 5.2 ± 0.1-fold (P = 2.4 × 10−3).
Overall, these results demonstrate that the Ig-like domain determines phage specificity, indicating that it functions as a sensor module within the pSlfn5 defence system. The acquired defence in the Sfo–Ror chimera shows that these domains are modular and can be exchanged between orthologues. However, the reduced phage defence of the chimera compared with RorSlfn5 suggests that effective immunity requires co-evolution between sensor and effector domains.
RorSlfn5 is a phage-activated tRNase
After identifying the viral cue that triggers pSlfn5 defence, we then investigated its mechanism. Co-expression of RorSlfn5 with its viral trigger led to growth arrest ~30 min after inducing T5.142 expression (Fig. 4a). This timing mirrors the kinetics of GFP expression under the same promoter, which became detectable 30 min post-induction (Fig. 4b). Control cells co-expressing RorSlfn5 and GFP or RorSlfn5E15A, D20A and T5.142 showed no growth arrest, demonstrating that it is mediated by T5.142-triggered nuclease activity of the pSlfn domain (Extended Data Fig. 5a,b).

a, Growth kinetics of MG1655 cells constitutively expressing RorSlfn5 with (0.2% arabinose (ara)) and without (0.2% glucose (gluc)) induction of T5.142 expression. b, Kinetics of GFP expression after induction of the pBAD promoter with arabinose. c, Schematics of the reporter assay for detecting SOS response upon DNA damage. d,e, Growth kinetics (d) and SOS-response reporter assay (e) of MG1655 treated with 100 nM mitomycin C or PBS. f, SOS-response reporter assay with RorSlfn5-expressing cells after induction of T5.142 expression. The kinetic assays in a–f were performed in four biological replicates. The data are shown as mean (centre line) ± s.d. (ribbon). g, Urea–PAGE of total RNA extracted from RorSlfn or RorSlfnE15A, D20A-expressing MG1655 at various timepoints after induction of T5.142 expression. Black arrows highlight RNA cleavage fragments. h, Schematics of the 3′-end RNA-seq approach used to map RorSlfn5-mediated RNA cleavage in T5 phage-infected cells. i, Total abundance of non-native (internal) 3′ ends in different RNA types in T5 phage-infected cells expressing RorSlfn5 or its nuclease-dead version (E15A and D20A mutation). The data are shown as the mean of three biological replicates ± s.d. Means were compared using a two-sided Welch’s t-test. The resulting P values were adjusted using the Holm method to account for multiple comparisons. j, The total abundance of internal 3′ ends in bacterial and viral tRNAs. The data are shown as the mean of three replicates.
Source data
To test whether RorSlfn5 targets DNA, we assessed bacterial DNA damage following T5.142-triggered RorSlfn5 nuclease activity. We used a fluorescent reporter plasmid (pSOS-GFP) that encoded a gfp gene under the control of a recA promoter, which is upregulated in the bacterial SOS response to DNA damage (Fig. 4c). Treatment of MG1655 with mitomycin C, a DNA-damaging agent, induced a strong fluorescent signal, which was absent in PBS-treated cells (Fig. 4d,e). By contrast, cells co-transformed with RorSlfn5, T5.142 and pSOS-GFP did not show increased fluorescence, suggesting that RorSlfn5 activation does not cause DNA damage and the SOS response (Fig. 4f and Extended Data Fig. 5c,d). These results indicate that RorSlfn5 does not act on DNA and probably retains RNase activity as its primary mechanism of toxicity, consistent with the function of human SLFN proteins3,4.
To test whether RorSlfn5 targets RNA, we co-transformed MG1655 cells with plasmids encoding the T5.142 trigger and either WT RorSlfn5 or a catalytically inactive mutant RorSlfn5E15A, D20A. We extracted total RNA from cells at 0, 30 and 60 min post-T5.142 induction. Urea–PAGE analysis of extracted RNA identified RNA fragments between 25 and 50 nt in size that appeared at 30- and 60-min post-induction in cells expressing RorSlfn5 but not the catalytically inactivated mutant, demonstrating that T5.142 expression triggers RorSlfn5-mediated RNA cleavage (Fig. 4g).
To map RNA cleavage induced by RorSlfn5, we performed RNA 3′-end sequencing (Fig. 4h). To validate this approach, we analysed RNA from E. coli expressing the PARIS-2 immune system. RNA sequencing (RNA-seq) revealed a significant enrichment of internal 3′ ends in tRNALys(UUU) (false discovery rate (FDR) of 0.002) in T5-infected cells expressing the active PARIS-2 defence compared with cells expressing the inactivated PARIS-2 system (AriBE26A mutant; Extended Data Fig. 6a–c). This result agrees with previous work demonstrating that activation of PARIS-2 immunity triggers AriB-mediated cleavage of tRNALys(UUU)20,33.
Next, we applied the same RNA-seq strategy to T5 phage-infected MG1655 cells expressing RorSlfn5 or its nuclease-dead mutant (Fig. 4h and Extended Data Fig. 6g). This analysis found no significant difference in the abundance of internal 3′ ends in messenger RNA (mRNA), non-coding RNA (ncRNA) or ribosomal RNA (rRNA), while internal tRNA 3′ ends were significantly enriched in cells expressing WT RorSlfn5 (Padj = 3.4 × 10−3; Fig. 4i). Analysis of tRNA mapping reads identified significant enrichment of total internal 3′-end counts in tRNALys(UUU) (194.1-fold, FDR of 2.4 × 10−4), T5 phage-encoded tRNAAla(UGC) (34.8-fold, FDR of 5.2 × 10−3) and tRNAMet(CAU) (145.4-fold, FDR of 4.4 × 10−5) in cells expressing WT RorSlfn5 compared with the nuclease-dead mutant (Slfn5dead; Fig. 4j and Supplementary Table 7). Position-specific analysis confirmed these enrichments and identified additional tRNAs with significant cleavage signatures, including tRNAVal(UAC), tRNAPhe(GAA) and tRNAIle(CAU) (FDR <0.05; Extended Data Fig. 6h). Together, these results indicated that RorSlfn5 preferentially cleaves tRNA upon phage infection, with tRNALys(UUU) fragments being most abundant in the cell.
Phage-activated RorSlfn5 cleaves the tRNA anticodon arm
We next sought to map RorSlfn5-mediated tRNA cleavage sites at single-nucleotide resolution. Mapping of 3′ ends in tRNALys(UUU) identified three highly enriched positions within the anticodon arm (29, 30 and 32), while the 5′-end mapping showed peaks at positions 39 and 41 (Fig. 5a,b). Correspondingly, sequence coverage across the entire anticodon loop of tRNALys(UUU) was absent, indicating complete removal of this region (Fig. 5c). A similar sequence coverage gap was observed between the 3′ and 5′ ends of tRNAMet(CAU) fragments (Extended Data Fig. 6i–k,m). This cleavage pattern suggests that RorSlfn5 may either excise the anticodon loop through two precise cuts or perform a single cut followed by trimming of the resulting fragments by host ribonucleases. Supporting the latter model, RNA-seq of cells expressing the PARIS immune system revealed a similar 11-nucleotide gap in the anticodon loop of tRNALys(UUU) spanning nucleotides 29 and 41, the latter position matching the AriB cut site identified in vitro20,33. These findings support a mechanism in which cleavage is followed by trimming of the anticodon loop (Extended Data Fig. 6d–f,l).

a,b, Position-specific mapping of internal 3′ ends (a) and 5′ ends (b) in tRNALys(UUU) in T5 phage-infected cells expressing RorSlfn5. The data are shown as the mean of three biological replicates. c, Alignment of sequencing reads to the lysW gene of MG1655 cells. One representative replicate from three biological replicates is shown. d, SEC of affinity-purified complex of RorSlfn5 and T5.142 (left). The elution fraction highlighted with a light blue rectangle was analysed with SDS–PAGE (right). The full SEC profile can be found in Extended Data Fig. 7. e, tRNA cleavage assays with 100 nM tRNALys(UUU) and 100 nM RorSlfn5 in the presence of T5.142 trigger (2 µM) and Mn2+ (2 mM). f, The same as in e but with 2 mM Mg2+, 2 mM Mn2+ or no metal and 10 mM EDTA. All shown assays were reproduced independently three times with similar results. g, Potential RorSlfn5 cut sites (triangles) in the anticodon arm of tRNALys(UUU), according to RNA-seq data in a and b. The red triangle shows the cut site supported by cleavage assays in e and f. h, A proposed model for RorSlfn5-mediated tRNA cleavage in vivo and subsequent host exoribonuclease (exoRNase) trimming of the anticodon loop.
Source data
To reconstitute tRNA cleavage in vitro, we expressed and purified RorSlfn5 and T5.142 proteins. The size-exclusion chromatography (SEC) profile of the RorSlfn5 (43.1 kDa) indicates that it assembles in higher-order oligomers, while T5.142 (13.8 kDa) eluted at a size consistent with a dimer or a trimer, based on gel filtration standards (Extended Data Fig. 7a,b). SEC with multi-angle light scattering (SEC–MALS) analysis confirmed this oligomerization, estimating an average mass of 261.0 ± 0.4 kDa, indicating that RorSlfn5 predominantly forms a hexamer at the tested conditions (Extended Data Fig. 7c). As Ig-like domains often mediate protein–protein interactions34, we hypothesized that the Ig-like domain of RorSlfn5 may detect phage by directly binding T5.142. To test this hypothesis, we co-expressed RorSlfn5dead with the T5.142 and performed a reciprocal affinity co-purification using a Twin-Strep-tag (Extended Data Fig. 7d). Both proteins co-purified from cell lysates regardless of which had the affinity tag and, after cleaving off the tags, RorSlfn5dead and T5.142 co-eluted in a single SEC peak, confirming a direct interaction and suggesting that binding of T5.142 triggers the tRNase activity of RorSlfn5 (Fig. 5d and Extended Data Fig. 7e).
Human SLFN nucleases catalyse tRNA cleavage using a manganese-dependent mechanism3,4. To test whether RorSlfn5 shares a similar mechanism, we incubated synthetic tRNALys(UUU) with purified RorSlfn5 and T5.142 in the presence of manganese, which yielded two major cleavage fragments sized between 25 and 50 nucleotides (Fig. 5e). Neither protein alone produced detectable cleavage and no activity was observed in the absence of metal or in the presence of EDTA (Fig. 5e,f). The detected tRNase activity was manganese specific as neither magnesium nor any other tested divalent metal supported cleavage in the tested conditions (Fig. 5f and Extended Data Fig. 8a). Mutations in the pSlfn active site (E15A, D20A) abolished cleavage activity, demonstrating that the Slfn nuclease domain of RorSlfn5 catalyses the cleavage reaction (Extended Data Fig. 8b). Upon titration of the trigger, tRNA cleavage was detectable with 1 µM T5.142 (100 nM RorSlfn5, 100 nM tRNALys(UUU)), while concentrations of T5.142 higher than 2 µM did not further enhance activity, suggesting that this trigger concentration saturates RorSlfn5 under the tested conditions (Extended Data Fig. 8c). Together, these cleavage assays demonstrate that RorSlfn5 functions as a manganese-dependent tRNase, activated by the phage tail assembly chaperone.
Testing additional tRNA substrates showed that bacterial tRNALys(UUU) and T5-encoded tRNAAla(UGC) were cleaved most efficiently, consistent with these tRNAs being the top hits in our RNA-seq data (Fig. 4j and Extended Data Fig. 8d). In vitro cleavage of tRNALys(UUU) produced two major products, consistent with a single cut (Fig. 5e,f), while the in vivo RNA-seq data identified two candidate RorSlfn5 cut sites at nucleotides 30–31 (site 1) and 39–40 (site 2) in tRNALys(UUU) (Fig. 5g). The fragment sizes observed in vitro support cleavage at predicted site 1 (30- and 46-nt products) over site 2 (37- and 39-nt products) (Fig. 5e,f). We therefore propose that in vivo RorSlfn5 cleaves the anticodon arm between nucleotides 30 and 31 (site 1), which is then followed by trimming of the anticodon loop by host ribonucleases (Fig. 5h).