
Synthesis and characterization of the conductive coordination nanozyme prodrug Cu–DHN
The conductive coordination nanozyme prodrug Cu–DHN was synthesized according to the protocol illustrated in Fig. 2a. Given that the enzymatic activity is critically dependent on the exposure ratio of active catalytic sites, smaller nanoparticles with higher specific surface areas generally exhibit enhanced POD through increased exposure of active sites.29 Inspired by the critical role of modulators in governing the growth of metal-organic frameworks (MOFs),30,31 we employed a modulator strategy using L-cysteine (Cys) to govern the coordination assembly process.

Engineering the architecture and coordination environment of the Cu–DHN nanozyme via L-Cysteine (Cys) modulation. a Synthesis scheme of Cu–DHN, illustrating the role of L-Cys as a coordination terminator and size regulator. The zoomed-in region illustrates the intermolecular hydrogen bonding between L-Cys molecules. This scheme was created with BioRender.com. b Hydrodynamic size distribution profiles (n = 3), and c TEM images of Cu–DHN with varying Cu:DHN:Cys feed molar ratios, revealing a non-monotonic size trend. d Variation profiles of hydrodynamic size for different M-DHN (M = Fe, Mn, Co) nanoparticles (n = 3) demonstrating the generalizability of Cys-mediated size control. e Fourier transform infrared (FT-IR) spectra confirming the involvement of phenolic –OH and thiol –SH groups; f normalized Cu K-edge XANES spectra, g Fourier transforms of k3-weighted Cu K-edge EXAFS spectra, and h high-resolution Cu 2p XPS spectra of Cu–DHN with varying Cu:DHN:Cys molar ratios, collectively evidencing the evolution of a mixed (O, S)-coordination shell and a rising Cu⁺/Cu²⁺ ratio with increasing Cys content. i HR-TEM image and corresponding SAED pattern, and j elemental distribution mapping and corresponding line-scan profile of Cu–DHN at a Cu:DHN:Cys molar ratio of 0.6:1:0.2. k XRD patterns, and l Brunauer–Emmett–Teller (BET) N2 adsorption–desorption isotherms demonstrating the pore structure evolution of Cu–DHN with varying Cu:DHN:Cys molar ratios, confirming the progressive surface passivation that defines the core–shell pore structure. Data are represented as mean ± SD
As shown in Fig. 2b, the hydrodynamic diameter of Cu–DHN decreased dramatically from 160 nm (Cu:DHN:Cys = 0.5:1:0, without Cys) to 70 nm when the molar ratio reached 0.6:1:0.2. However, further increasing the Cys content to craft Cu–DHN (Cu:DHN:Cys = 0.75:1:0.5) and Cu–DHN (Cu:DHN:Cys = 1:1:1) enlarged the particles to 110 and 140 nm, respectively. Thus, we demonstrated that L-Cys served as a potent molecular modulator to precisely control the size of Cu–DHN, a critical parameter for optimizing its peroxidase (POD)-mimetic activity. This size control was achieved by tuning the competition between infinite coordination polymerization and Cys-mediated termination, with excessive Cys inducing a reversal of size reduction guided by crystallization due to intermolecular hydrogen bonding. The efficacy of this modulation is directly evidenced by the nonmonotonic variation in nanoparticle dimensions.
The parent polyphenol ligand 1,5-DHN, featuring two phenolic hydroxyl groups, promotes uncontrolled polymer growth by coordinating with Cu2+ ions. The initial coordination at the 1-position creates steric hindrance, forcing the 5-position hydroxyl to coordinate with another Cu2+, leading to uncontrolled growth of infinite coordination polymers and larger nanoparticles. The introduction of Cys, possessing a single thiol group, effectively terminated this infinite coordination assembly, thereby restricting particle growth and yielding smaller nanoparticles.30 However, excessive Cys triggered hydrogen bond-mediated self-assembly between Cys molecules,32 counteracting the size-reduction effect. At higher concentrations, the excess free Cys in the synthesis solution (at pH ≈ 8) predominantly existed in a zwitterionic form. This facilitates the formation of robust intermolecular hydrogen bonds between the NH3+ and COO− groups and may further be stabilized by disulfide-linked L-cystine formation. This hydrogen bond-mediated self-assembly and crystallization of Cys acts as a secondary aggregation force, counteracting the primary size-reduction effect and leading to the observed increase in hydrodynamic diameter. Multifaceted characterization robustly confirmed the precision, universality, and stability of Cys-mediated size control. This strategy not only yielded optimally sized nanoparticles for our Cu-based metal-phenolic network but also proved applicable across a spectrum of catalytic metals, underscoring its potential as a general platform for nanozyme design.
Complementary TEM and SEM characterization (Fig. 2c, Supplementary Fig. 3) confirmed these spherical-like morphologies of all nanostructures and validated the size trends observed by DLS. The pristine Cu–DHN (0.5:1:0) exhibited a ~150 nm diameter, while Cys-optimized samples (0.6:1:0.2) showed a dramatic size reduction to ~50 nm. Subsequent Cys overloading (0.75:1:0.5 and 1:1:1) progressively increased particle sizes to 100 and 120 nm, respectively, demonstrating precise size modulation through stoichiometric control. The optimal Cu:DHN:Cys = 0.6:1:0.2 achieves a critical balance between leashing infinite coordination polymerization and preventing hydrogen bond-driven agglomeration. The slight discrepancy between the smaller dry-state (TEM/SEM) and larger hydrodynamic (DLS) diameters is expected, attributable to the thin hydration layer surrounding the nanoparticles in solution.
Furthermore, we extended the synthesis to three additional POD-active metal ions (Fe3+, Mn2+, and Co2+) for coordination coassembly with DHN and Cys (Fig. 2d). Dynamic light scattering (DLS) analysis revealed that while the absolute particle sizes of M-DHN (M = Fe, Mn, Co) varied depending on the metal ion species, the nonmonotonic size variation pattern in response to changing Cys composition remained consistent across all systems. Moreover, Mg–DHN (Mg:DHN:Cys = 0.6:1:0.2), Cu–Cys (Cu:DHN:Cys = 0.5:0:1, without DHN), and the parent drug group Cu–juglone (Cu:juglone:Cys = 0.6:1:0.2) were synthesized as control materials for further evaluation (Supplementary Fig. 4). Multifaceted characterization robustly confirmed the precision, universality, and stability of Cys-mediated size control. This strategy not only yielded optimally sized nanoparticles for our Cu-based metal-phenolic network but also proved applicable across a spectrum of catalytic metals, underscoring its potential as a general platform for nanozyme design. Dispersing the prodrug nanoparticle Cu–DHN in various media (Supplementary Fig. 5) demonstrated that it largely maintained its integrity and uniformity, with only a slight size reduction observed in acidic environments (pH < 5), indicating good colloidal stability for subsequent biological applications.
Subsequently, we investigated the driving forces behind the coordination coassembly of Cu–DHN. Initial UV‒vis absorption spectral analysis revealed characteristic peaks of DHN in Cu–DHN (Supplementary Fig. 6). Notably, broad-band absorption in the infrared region was observed, attributed to enhanced conjugation resulting from J-aggregation of the DHN assembly,33 suggesting successful coordination between DHN and Cu2+ ions. Fourier transform infrared spectroscopy (FT-IR) analysis demonstrated significant attenuation of the phenolic hydroxyl group’s in-plane bending vibration at 1421 cm−1 in DHN following Cu2+ coordination (Fig. 2e). Concurrently, the characteristic S–H stretching vibration of Cys at 2646 cm−1 disappeared after Cu2+ interaction, indicating restricted vibrational modes of both phenolic hydroxyl and thiol groups upon metal coordination. These observations preliminarily confirm the coassembly of Cu2+ with phenolic hydroxyl groups of DHN and thiol groups of Cys, leading to the formation of a Cu–DHN conductive coordination nanozyme prodrug. Our comprehensive spectroscopic analysis also revealed that L-Cys orchestrated a multifunctional coordination network for optimized nanozyme performance. Apart from being a mere comonomer, it critically governs the coordination geometry, modulates the copper valence state, and ultimately tailors the prodrug’s catalytic potency and therapeutic potential. This integrated role is fundamental to achieving both controlled nanoparticle size and high catalytic activity.
To further elucidate the coordination mechanism, we employed XPS and synchrotron radiation characterization. Beyond simple coordination, L-Cys actively engineered a hybrid Cu⁺/Cu²⁺ interface, which is a kernel for catalytic activation. XPS and synchrotron radiation analyses provided atomic-level insights into this sophisticated mechanism. EXAFS data unambiguously confirmed the coexistence of Cu–O (from DHN) and Cu–S (from Cys) coordination shells (Fig. 2f, g). X-ray spectroscopy (XPS) analysis revealed a 1.33 eV negative shift in the O 1s binding energy for DHN after Cu2+ coordination, while a 0.27 eV positive shift occurred in the S 2p binding energy for Cys (Supplementary Fig. 7), suggesting a distinct electron cloud distribution in the Cu–O and Cu–S bonds. Fourier transforms of the k3-weighted Cu extended X-ray absorption fine structure (XAFS) provided direct evidence of coordination environments (Fig. 2g): Cu–DHN (Cu:DHN:Cys = 0.5:1:0) exhibited a dominant peak at 1.56 Å corresponding to Cu-–O coordination shells, consistent with the CuO reference. Conversely, Cu–Cys showed a characteristic Cu–S coordination peak at 1.80 Å, confirming a heterogeneous coordination environment with both oxygen and sulfur atoms in the first shell (Supplementary Table 2).
In addition, progressive increases in Cys content (Cu:DHN:Cys from 0.6:1:0.2 to 1:1:1) resulted in intermediate first-shell radii between the Cu–O and Cu–S configurations, confirming the coexistence of both coordination modes (Supplementary Fig. 8, Supplementary Table 2). Cu K-edge X-ray absorption near-edge structure (XANES) spectra revealed significant valence state variations: an incremental shift occurred from Cu2+ toward Cu+ as the Cys content increased (Fig. 2f). The Cu–DHN (Cu:DHN:Cys = 0.5:1:0) exhibited Cu K-edge characteristics matching CuO, indicating the exclusive presence of Cu2+. Increasing the Cys content progressively shifted the absorption edge toward the Cu2S reference, with Cu–Cys displaying an intermediate edge position between CuS and Cu2S, suggesting a hybrid of Cu+/Cu2+ valence states in Cys-containing Cu–DHN. XPS deconvolution of Cu 2p peaks quantified this valence transition: Cu+ content increased from 14% to 70% as the Cys ratio increased from 0.2 to 1 equivalent (Fig. 2h, Supplementary Fig. 9). This was a direct consequence of the reducing capacity of Cys, which reduced a portion of the Cu2+ precursors to Cu+ during synthesis. This dual valency was proven to be a cornerstone of our design: it ensured storage stability as a Cu2+-based coordination polymer, while preloading the nanoparticle with a significant surface reservoir of the more bioactive Cu+. The redox-mediated valence modulation is rooted in the reducing capacity of Cys, which reduces Cu2+ to Cu+ while terminating nanoparticle growth, resulting in surface-enriched Cu+ species. The surface-preloaded Cu+ species endow Cu–DHN with dual therapeutic advantages: (i) facile catalytic activation for rapid •OH generation from H2O2 upon cellular internalization34 and (ii) enhanced cuproptosis induction through elevated Cu+ levels.26
In this process, Cys plays three indispensable roles. As a size regulator, it terminated the infinite coordination polymerization by surface-limited coordination; as a coordination partner, it created a mixed (O, S)-ligand field; as a redox modulator, it generated a stable yet Fenton reaction-ready Cu+/Cu2+ hybrid state. This sophisticated coordination design yields a prodrug that is stable in storage yet primed for rapid catalytic activation and cuproptosis induction in the tumor microenvironment, perfectly aligning the synthesis strategy with the therapeutic objective.
To elucidate the mechanism underlying Cys-regulated particle size control in Cu–DHN, we conducted systematic structural and compositional analyses. Our structural analyses established that L-Cys governed the particle size by terminating the uncontrolled coordination growth of the copper–polyphenol framework Cu–DHN, leading to the formation of a unique crystalline core–amorphous shell structure. High-resolution TEM (HR-TEM) imaging combined with elemental mapping of Cu–DHN (Cu:DHN:Cys = 0.6:1:0.2) revealed distinct structural features. Elemental line scanning and mapping of Cu–DHN (Cu:DHN:Cys = 0.6:1:0.2) provided critical evidence for this mechanism. Elemental mapping and line scanning revealed that sulfur (S) signals were predominantly localized at the particle surface, whereas Cu and oxygen (O) were distributed uniformly throughout the core–shell architecture (Fig. 2i). This visual evidence echoed the mechanism where initial, unrestricted coordination between the bidentate phenolic hydroxyl groups of DHN and Cu2+ forms the crystalline core, while subsequent surface termination by monodentate Cys creates the disordered shell (Fig. 2j). This spatial segregation confirmed that Cys-derived sulfur participates in surface coordination without penetrating the crystalline core.35 The intact crystalline core region (evidenced by clear lattice fringes and diffraction spots) was encapsulated by a loose, amorphous outer shell. This spatial segregation confirms that Cys does not incorporate into the growing crystal lattice but rather coordinates with Cu2+ at the particle interface, effectively passivating the surface and arresting further growth.
XRD and BET analyses unraveled this core–shell model and excluded a simple physical mixture. XRD patterns further corroborated the core–shell configuration (Fig. 2k). Cu–DHN (Cu:DHN:Cys = 0.5:1:0) exhibited characteristic diffraction peaks at 7.07°, 12.94°, 15.38°, 20.33°, 23.84°, 25.21°, 26.20°, and 36.36°, consistent with the HR-TEM observations. Progressive Cys incorporation (0–1 equivalent) diminished the diffraction peaks of the crystalline Cu–DHN core, recognized by gradual attenuation of Cu–DHN diffraction intensities and emergence of Cu–Cys signatures at 18.90°, 28.49°, 33.15°, and 34.49°. Complete suppression of Cu–DHN peaks occurred at the highest Cys ratio, where Cu:DHN:Cys = 1:1:1. However, physical mixtures of Cu–DHN (Cu:DHN:Cys = 0.5:1:0) and Cu–Cys maintained distinct and distinguishable diffraction patterns (Supplementary Fig. 10). This critical comparison confirmed that rather than being physically blended, the Cys-derived shell was chemically bound, forming a conformal coating that shielded the core from X-ray detection.36
BET analysis revealed pore volume variations supporting the proposed model (Fig. 2l). Compared to Cu–Cys (0.085 cm³/g), Cu–DHN exhibited a significantly higher pore volume (0.238 cm³/g), reflecting its larger pore dimensions favorable for gas diffusion. This discrepancy confirmed that the surface Cu–Cys coating acted as a molecular sieve, physically restricting gas access to the porous Cu–DHN core. In this regard, increasing Cys content progressively reduced measured pore volumes below theoretical values calculated for physical mixtures, with the deviation amplifying at higher Cys ratios (Supplementary Table 3). This phenomenon suggests that conformal Cu–Cys surface coatings restrict gas access to the porous Cu–DHN core through size-exclusion effects.37
The superior catalytic performance of Cu–DHN arises from synergistic size control and intrinsic electron shuttling
Initial performance screening to decode the principles and outcomes of size optimization is necessary. Subsequently, we demonstrated that the superior peroxidase (POD)-like activity and glutathione peroxidase (GSH-Px)-like activity of our nanozyme emerged from the sophisticated interplay of a minimized particle size, an optimized Cu+/Cu2+ valence state, and a conductive polyphenol backbone that enables efficient electron shuttling. As shown in Fig. 3, this triad collaborated to maximize active-site exposure, facilitate the crucial Fenton redox cycle, and engage the entire nanoparticle volume in catalysis.

The electron-shuttling architecture of Cu–DHN underpins its superior and sustained catalytic activity. a GSH-Px-like activity evaluation of Cu–DHN with varying Cu:DHN:Cys feed molar ratios via DTNB assay. The column chart presents the peak values of UV–Vis absorbance at 412 nm. b and c POD-like property of Cu–DHN with varying Cu:DHN:Cys molar ratios via (b) TMB colorimetric assay (the column chart presents the peak values of UV–Vis absorbance at 662 nm), and c ESR analysis using DMPO as the spin trapping agent, confirming •OH as the primary reactive species. d Catalytic Hill plots of Cu–DHN with varying Cu:DHN:Cys molar ratios in the presence of GSH using H2O2 as substrate. e, f Direct evidence for the electron-shuttling mechanism illustrated by high-resolution Cu 2p XPS, presenting highly efficient Cu⁺/Cu²⁺ cycling in e the agile behavior of Cu–DHN (Cu:DHN:Cys = 0.6:1:0.2) and the retarded behavior of f Cu–Cys. g Schematic representations of electron transport via a conductive DHN-promoted pathway. This scheme was created with BioRender.com. h Electrical conductivity of Cu–DHN with varying Cu:DHN:Cys molar ratios. Data are represented as mean ± SD (n = 3). i Time-dependent MB degradation process after incubation with Cu–DHN (Cu:DHN:Cys = 0.6:1:0.2) in the presence of GSH and H2O2. j Juglone yield from Cu–DHN with varying Cu:DHN:Cys molar ratios after GSH and H2O2 incubation over time (n = 3). Data are represented as mean ± SD
It has been reported that Cu2+-based nanozymes can oxidize GSH to generate glutathione disulfide (GSSG) while being reduced to Cu⁺ with POD activity, thereby disrupting redox homeostasis in tumor cells.23 Therefore, the GSH-Px activity of Cu–DHN was quantitatively assessed using Ellman’s colorimetric assay probed by DTNB.38 As shown in Supplementary Fig. 11, Cu–DHN (Cu:DHN:Cys = 0.6:1:0.2) exhibited a time-dependent GSH-depletion capacity. Subsequent comparative analysis of Cu–DHN with different Cu:DHN:Cys feed ratios revealed that Cu–DHN (Cu:DHN:Cys = 0.6:1:0.2) demonstrated optimal GSH-depletion efficiency (Fig. 3a). This enhancement is attributed to its minimized particle size (approximately one-third that of Cu–DHN with Cu:DHN:Cys = 0.5:1:0), collectively yielding a 27-fold increase in specific surface area compared to Cu DHN (Cu:DHN:Cys = 0.5:1:0). The expanded surface area facilitates the exposure of catalytically active sites, thereby promoting interfacial redox reactions with GSH (Supplementary Fig. 12).
The POD activity was further investigated through TMB chromogenic assays measuring •OH generation from the dismutation of H2O2 catalyzed by Cu–DHN and Cu–Cys with varying Cu:DHN:Cys feeding ratios.39 Kinetic analysis demonstrated a negative correlation between nanoparticle size and •OH production, with smaller Cu–DHN (Cu:DHN:Cys = 0.6:1:0.2) exhibiting superior catalytic performance due to increased active-site accessibility (Fig. 3b). The type of ROS was identified by electron spin resonance (ESR) spectroscopy. DMPO probing presented characteristic 1:2:2:1 quartet signals, confirming •OH as the predominant species contributing to catalytic activities (Fig. 3c, Supplementary Fig. 13). Kinetic analysis fitting the Hill equation revealed that Cu–DHN’s maximum reaction rate (Vmax) inversely correlated with particle size and consistently surpassed all controls (Fig. 3d), including previously reported nanozymes (Supplementary Table 4). The distinct kinetic phases can be rationalized by the initial scavenging of •OH by GSH and DHN moieties, followed by a rapid acceleration phase once •OH generation outstrips consumption. This efficient kinetics was a direct consequence of the high-density, readily accessible active sites enabled by the electron-conducting framework.
Notably, despite comparable particle sizes between Cu–Cys and Cu–DHN (Cu:DHN:Cys = 0.6:1:0.2), the former exhibited significantly inferior GSH-depletion performance. Cu–DHN outperformed Cu–Cys by 3.2-fold in •OH yield despite comparable dimensions. As anticipated, minimizing the particle size to ~70 nm (Cu:DHN:Cys = 0.6:1:0.2) dramatically enhanced both GSH-depletion and •OH generation, owing to the maximized specific surface area. Nevertheless, the critical limitation of relying solely on surface atoms was challenged by the direct comparison of Cu–DHN (Cu:DHN:Cys = 0.6:1:0.2) with Cu–Cys: despite comparable sizes, Cu–DHN outperformed Cu–Cys by a significant margin in both GSH consumption and •OH yield. This disparity suggested that additional factors beyond particle size optimization contribute to the enhanced catalytic activity. Thus, we hypothesized that the conjugated backbone of DHN was critical for unlocking a robust and sustainable catalytic function. This “electron highway” of the π-conjugated structure not only drove superior enzyme kinetics but also facilitated a complete prodrug activation cycle, setting Cu–DHN apart from its counterparts. Consequently, we quantitatively captured the properties of this “electron highway” using XPS and four-point probe conductivity measurements.
Upon GSH incubation, XPS showed that in Cu–DHN, the Cu⁺ proportion surged from 14% to 81% (Fig. 3e), indicating a highly efficient and profound reduction process that engaged the nanoparticle core. Subsequent H2O2 exposure triggered near-complete (90%) reoxidation to Cu²⁺ (Fig. 3f), demonstrating a robust and reversible redox cycle essential for sustained •OH generation. In stark contrast, Cu–Cys showed lethargic valence changes (<20% variation), confirming its confinement to superficial, limited reactions (Supplementary Fig. 14). Conductivity measurements provided further evidence of enhanced charge transfer in Cu–DHN, showing significantly higher conductivity than Cu–Cys (Fig. 3h). This phenomenon was a direct result of the synergistic effects between efficient Cu+/Cu2+ valence transitions and DHN-mediated electron transport through the nanoparticle’s core–shell architecture (Fig. 3g). Postreaction characterization via TEM, DLS, and XRD confirmed that structural integrity was maintained with preserved crystallinity and consistent size distribution trends (Supplementary Figs. 15, 16), indicating that the GSH/H2O2 treatment had a negligible influence on the particle size and structure of Cu–DHN. This compelling evidence substantiates the superior efficacy of our dual strategy combining particle size optimization and electron transfer engineering for POD performance enhancement.
Methylene blue (MB) bleaching assays confirmed persistent •OH generation over 180 min (Fig. 3i). Critically, this sustained radical flux efficiently oxidizes the coordinated DHN into its active anticancer form, juglone. Distinct DHN-to-juglone conversion efficiencies were mediated by Cu–DHN with varying Cu:DHN:Cys ratios. After 3 h of coincubation, Cu–DHN (Cu:DHN:Cys = 0.6:1:0.2) achieved ~55% DHN oxidation to juglone, whereas Cu–DHN (Cu:DHN:Cys = 0.5:1:0) exhibited a substantially lower conversion efficiency (~30%) under identical conditions (Fig. 3j). Notably, the juglone conversion rates across Cu–DHN at different Cu:DHN:Cys ratios showed a positive correlation with their respective POD enzymatic activities. This proportional relationship conclusively linked the superior catalytic properties of Cu–DHN directly to its therapeutic function. To confirm that the observed therapeutic effects were due to the Cu-dependent catalytic activity, we synthesized a control material by replacing the active center with redox-inert Mg2+ ions and forming Mg–DHN (Mg:DHN:Cys = 0.6:1:0.2). The complete catalytic inertia (Supplementary Fig. 17) of the redox-inert Mg–DHN control, which exhibited negligible activity across all assays, underscored that the observed therapeutic effects are specifically due to the Cu-dependent catalytic activity, providing a rigorous negative control.
In conclusion, the orchestrated interplay of size reduction, a Cu⁺/Cu²⁺ hybrid valence state, and a conjugated ligand backbone is paramount. Particularly, the electron highway within Cu–DHN has done more than just enhance kinetics; it ensured a durable catalytic performance that powers the continuous on-site production of both cytotoxic radicals and the activated chemotherapeutic agent, solidifying its design as an integrated and self-sufficient prodrug system.
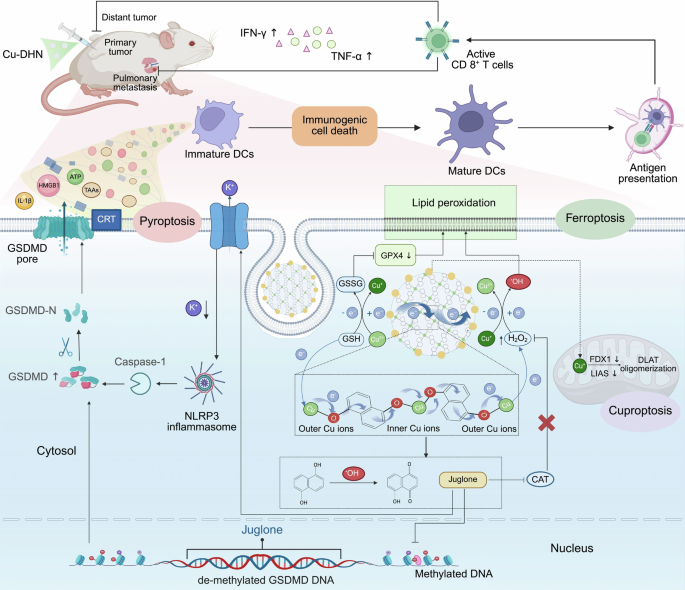
Cu–DHN elicits potent antitumor immunity via a synergistic cascade of pyroptosis-dominant programmed cell death
Cu–DHN orchestrated a potent and tumor-specific therapeutic outcome by sequentially activating a cascade of pyroptosis, cuproptosis, and ferroptosis, with pyroptosis serving as the dominant pathway. This tripartite cell death synergy not only effectively killed cancer cells but also, crucially, initiated powerful immunogenic cell death (ICD) to stimulate an in situ vaccination effect, as shown in Fig. 4.

In vitro effects of Cu–DHN (Cu:DHN:Cys = 0.6:1:0.2) on 4T1 cells. a CLSM images and b mean fluorescence intensity (MFI) of 4T1 cells stained by DCFH-DA (ROS probe) after co-incubation with Cu–DHN for 6 h (n = 3). c Juglone yield in 4T1 cells after co-incubation with Cu–DHN for 12 h (n = 3). d Cell viability curves of 4T1 cells after co-incubation with Cu–DHN for 24 h (n = 6). e Bright-field microscopic images of 4T1 cells after co-incubation with Cu–DHN for 18 h (yellow arrows highlight membrane-bound vesicular protrusions indicative of pyroptotic morphology). f Intracellular K+ content in 4T1 cells after co-incubation with Cu–DHN for 6 h (n = 3). g NLRP3 content, h Caspase-1 activity, i GSDMD-FL content, j GSDMD-N content in 4T1 cells after co-incubation with Cu–DHN for 18 h (n = 3). k IL-1β secretion from 4T1 cells after co-incubation with Cu–DHN for 18 h (n = 3). l Internalized amounts of Cu by 4T1 cells after co-incubation with Cu–DHN for 6 h (n = 3). m FDX1 content in 4T1 cells after co-incubation with Cu–DHN for 18 h (n = 3). n GPX4 activity in 4T1 cells after co-incubation with Cu–DHN for 18 h (n = 3). o CLSM images of 4T1 cells stained by Liperfluo (LPO probe) after co-incubation with Cu–DHN for 12 h (n = 3). p Flow cytometry analysis of 4T1 cells labeled by AF488-CRT antibody after co-incubation with Cu–DHN for 18 h. Release of q HMGB-1, and r ATP from 4T1 cells after co-incubation with Cu–DHN for 18 h (n = 3). s BMDC maturation after co-incubation with Cu–DHN treated 4T1 cells for 24 h (n = 3). Cu concentration: 0.020 mM, DHN or juglone concentration: 0.033 mM. Data are represented as mean ± SD
The cellular uptake and lysosomal escape ability of Cu–DHN were evaluated first. As shown in Supplementary Fig. 18a, intracellular Cu–DHN accumulation exhibited a significant time-378-dependent increase during 6 h of coincubation, reaching saturation thereafter. Moreover, Cu–DHN exploits macropinocytosis for entry and triggers a self-limiting cascade of lysosomal destabilization (Supplementary Fig. 18b, 19). Afterwards, general intracellular •OH generation was probed by DCFH-DA via confocal laser scanning microscopy (CLSM) (Fig. 4a, b). Both the free CuCl2 and its coadministration groups with juglone and DHN exhibited significantly weaker DCF green fluorescence intensity compared to the coordinated composites Cu–DHN and Cu–juglone. This disparity arose from enhanced POD-like activity in Cu–DHN through particle size optimization and conductive structure modulation. In contrast, Cu–Cys showed significantly reduced •OH generation compared to Cu–DHN, since the absence of “redox capillaries” in ligands hampered efficient electron transfer and impeded the swift revival of catalytic activity. This •OH not only directly caused damage but also oxidized the prodrug DHN into juglone.
The •OH generation mechanism requires both catalysts and substrates, and the superior POD activity of Cu–DHN enables efficient •OH generation under sufficient H2O2 conditions.24 In this established self-amplifying loop, juglone, oxidized from DHN by •OH, inhibits catalase (CAT) activity (Supplementary Fig. 20), thereby elevating intracellular H2O2 to boost further •OH generation and DHN oxidation. Based on the exceptional •OH-generating capacity of Cu–DHN, we quantified the intracellular DHN-to-juglone conversion. CuCl2 + DHN combinations oxidized merely 5% DHN, while Mg–DHN nanoparticles (lacking POD activity) showed negligible conversion. In contrast, Cu–DHN achieved >50% DHN oxidation after 12 h of incubation (Fig. 4c), attributable to its robust •OH production.
Then, we established the superior and selective cytotoxicity of Cu–DHN. Cu–DHN exhibited micromolar-level IC50 values (IC50 = 0.026 mM Cu and 0.042 mM DHN) against 4T1 tumor cells, outperforming control nanozymes such as Cu–Cys (IC50 = 0.067 mM Cu) and matching the potency of the active parent drug version Cu–juglone (IC50 = 0.016 mM Cu, 0.027 mM juglone) (Fig. 4d). Crucially, this potency was tumor-selective: unlike the indiscriminate toxicity of the parent drug Cu–juglone (Supplementary Table 5), Cu–DHN maintained high viability in normal cells, demonstrating its successful activation only within the tumor’s unique redox environment (Supplementary Fig. 21). Cu–DHN maintained >75% viability in HUVECs, NIH 3T3 cells, and HSF normal cells at 0.1 mM DHN compared with Cu–juglone (<35% viability at 0.08 mM juglone) (Supplementary Fig. 21a). This specificity was governed by a tumor-selective GSH “AND” H2O2 logic gate and a self-amplifying catalytic loop: the •OH-generated juglone inhibited catalase, elevating intracellular H₂O₂ to fuel further •OH production and DHN-to-juglone conversion (>50% in 12 h), a process inaccessible to less catalytic analogs (Supplementary Fig. 21b).
In 4T1 cells, which are inherently gasdermin deficient,40 Cu–DHN successfully initiated pyroptosis, as evidenced by the formation of characteristic pyroptotic bodies (Fig. 4e, Supplementary Figs. 2, 22). Mechanistically, it activated the canonical pathway: K⁺ efflux triggered NLRP3 inflammasome assembly and Caspase-1 cleavage (Fig. 4f-h). Most importantly, Cu–DHN epigenetically upregulated GSDMD transcription through DNA methyltransferase inhibition (Fig. 4i, Supplementary Figs. 23a, b, 24), thereby “priming” low-GSDMD tumor cells for pyroptosis. This ensured the cleavage of GSDMD and the mature release of IL-1β through the resulting pores (Fig. 4j, k). This dual-pathway activation (epigenetic directing and functional reactivation) overcomes intrinsic resistance in GSDMD-low tumors while avoiding off-target pyroptosis risks, ensuring robust pyroptosis even in challenging models.
Beyond pyroptosis, our platform ingeniously overcomes a central paradox in cuproptosis therapy. While copper ions are potent inducers of cuproptosis, their efficacy is severely impeded by the cell’s sophisticated homeostatic mechanisms that minimize free copper flux.26 However, our design resolves this by exploiting macropinocytosis—a rampant endocytic pathway in cancer cells—to deliver copper as a coordinated polymer nanoparticle. As quantitatively demonstrated by our earlier ICP-MS uptake assays (Fig. 4l), the encapsulation of copper within a coordination polymer framework shifts the primary cellular entry pathway from regulated, low-capacity transporter-mediated uptake to efficient, nonspecific macropinocytosis. This paradigm shift in delivery mechanics was evidenced by the dramatically enhanced cellular copper accumulation achieved by all our nanoformulations (regardless of Cu–DHN, Cu–juglone, and Cu–Cys) compared to unbound CuCl2. This strategy bypassed physiological copper regulators and ensured a massive, uncontrolled intracellular delivery of copper.
Since macropinocytosis is a gateway that is particularly active in nutrient-demanding cancer cells, the preactivated Cu⁺ species within the Cu–DHN particle readily engage the cuproptosis pathway, leading to the marked downregulation of FDX1 and LIAS (Fig. 4m, Supplementary Fig. S25c). Thus, the same cellular uptake mechanism that underlies its dominant pyroptosis effect also empowers a potent (Supplementary Figs. 23c, d, 24a, b), synergistic cuproptosis, making Cu–DHN a comprehensive cell death inducer. Rescue experiments demonstrated that the contribution of cuproptosis is secondary to that of pyroptosis. Copper sequestration by the copper chelator TTM illustrated the central role of Cu–DHN as a motor of cuproptosis (Supplementary Fig. 25). Apart from pyroptosis and cuproptosis, Cu–DHN triggered lipid peroxidation by depleting GSH and disabling GPX4, initiating ferroptosis (Fig. 4n, o). The MDA concentration differentiated the GSH-Px-dependent ROS storm from the general oxidative attack. However, rescue experiments confirmed that the contribution of ferroptosis was subdominant (Supplementary Fig. 26), as its inhibition failed to fully reverse cell death.
As comprehensively detailed in Fig. 4, Cu–DHN elicited potent immunogenic cell death in 4T1 cells by synchronously activating pyroptotic, cuproptotic, and ferroptotic pathways. Flow cytometry analysis demonstrated that Cu–DHN induced a superior immunogenic cell death (ICD) effect, with CRT exposure reaching ~90% (Fig. 4p, Supplementary Fig. 23). This effect was complemented by HMGB-1 and ATP secretion levels comparable to those of the parent drug Cu–juglone, all of which were significantly higher than the levels observed in other control groups. Through massive TAA and DAMP release (Fig. 4q, r), the endogenous cancer vaccine formation mechanism revealed Cu–DHN as a critical ICD inducer comparable to Cu–juglone, showing 2-fold higher DC maturation rates than other control groups (Fig. 4s, Supplementary Fig. 28) and effectively priming a tumor-specific T-cell response. Notably, the DC maturation induced by pyroptosis-primed cells was 2-fold higher than that from cells undergoing only cuproptosis/ferroptosis, underscoring pyroptosis as the principal driver of ICD.
In vivo antitumor efficacy and mechanism of Cu–DHN
To validate that our nanozyme prodrug Cu–DHN executes its designed therapeutic mechanisms within the complex tumor microenvironment, we first employed an orthotopic 4T1 breast cancer model with local administration (Fig. 5a). Cu–DHN treatment significantly suppressed orthotopic tumor growth over 14 days, demonstrating comparable inhibition to the Cu–juglone group and superior to all controls (Fig. 5b, Supplementary Fig. 29). This potent antitumor activity of Cu–DHN translated into a 75% tumor growth inhibition (TGI) (Fig. 5c,d), significantly reduced tumor weights, and a notable survival benefit. The potent antitumor activity translated into significant survival prolongation in Cu–DHN-treated tumor-bearing mice relative to control groups (Fig. 5e). Critically, the Mg–DHN control, lacking catalytic activity, was indistinguishable from the PBS group, underscoring that the therapeutic effect is contingent on metal-dependent catalysis. Furthermore, the mixture of free CuCl2 + DHN showed minimal efficacy, attributable to its inadequate cellular internalization and insufficient ROS production for prodrug activation, highlighting the indispensable role of nanocoordination in ensuring codelivery and activation.

In vivo effects of Cu–DHN (Cu:DHN:Cys = 0.6:1:0.2) on 4T1 tumor. a Schematic illustration of inhibiting tumor growth via inducing pyroptosis, cuproptosis, and ferroptosis by a single intratumoral injection of Cu–DHN. b Longitudinal monitoring of tumor growth following single-dose Cu–DHN intratumoral administration (3.78 μmol kg−1 Cu, 6.24 μmol kg−1 DHN or juglone) (n = 5). c Weights of dissected tumors, and d tumor inhibition rate at 14 days post-treatment (n = 5). e Survival rate analysis of 4T1 tumor-bearing BALB/c mice treated with Cu–DHN (n = 8). f CLSM images and MFI of tumor slices labeled with g DHE (1 day post-treatment), h GSDMD-N antibody (14 days post-treatment), i FDX1 antibody (14 days post-treatment), j Liperfluo (1 day post-treatment), and H&E (14 days post-treatment) (n = 3). Data are represented as mean ± SD
Histopathological analysis of excised tumors revealed the molecular basis for Cu–DHN’s efficacy (Fig. 5f–j): a concerted attack via multiple programmed cell death pathways. We observed (i) the execution of pyroptosis, evidenced by upregulated GSDMD-N; (ii) the induction of cuproptosis, marked by downregulated FDX1; and (iii) the triggering of ferroptosis, demonstrated by elevated lipid peroxidation (LPO) levels. This multimodal cell death signature, corroborated by the marked reduction in tumor cellularity in H&E-stained cells, confirms that Cu–DHN’s catalytic activity successfully translates into a powerful and complementary therapeutic outcome in vivo.
Thus far, local administration of Cu–DHN has conclusively validated our design principle: the nanozyme efficiently generates •OH in the tumor milieu, activates the DHN-to-juglone conversion, and subsequently unleashes a potent, synergistic combination of pyroptosis, cuproptosis, and ferroptosis. The fact that Cu–DHN, a stable prodrug, matches the efficacy of the preformed toxic agent (Cu–Juglone) is definitive proof of its successful in vivo activation and mechanistic fidelity.
This robust foundational efficacy and mechanism validation upon local delivery strongly supports its further investigation under more clinically relevant, systemic administration routes. We transitioned from local (intratumoral) administration to a clinically relevant systemic (intravenous) delivery model (Supplementary Fig. 30a). Our results demonstrate that low-dose Cu–DHN exhibited relatively weak tumor growth inhibition, whereas moderate- and high-dose Cu–DHN significantly suppressed tumor progression. Moreover, compared to the high-dose group, the moderate-dose regimen did not cause significant body weight loss in tumor-bearing mice. Thus, moderate-dose Cu–DHN demonstrates both potent antitumor efficacy and favorable biosafety. (Supplementary Fig. 30b, c).
Pharmacokinetic evaluation revealed that moderate-dose Cu–DHN achieves a serum half-life of 2.01 h (Supplementary Fig. 30d), a performance closely comparable to PEGylated nanomaterials as reported. Biodistribution analysis confirmed the success of this strategy, showing significant tumor enrichment of Cu–DHN, peaking at 18.37% ± 1.89% ID g−1 at 12 h postinjection (Supplementary Fig. 30e).
The cornerstone of clinical translation is systemic safety. Beyond direct cancer cell killing, Cu–DHN orchestrated a potent systemic immune response. ELISA analyses revealed marked elevations in key antitumor cytokines, including TNF-α and IFN-γ, which peaked between day 3 and day 7 posttreatment. The subsequent slight decline reflects a natural and desirable immune regulatory process, preventing potential immune-related adverse events (irAEs) and indicating well-controlled activation (Supplementary Fig. 30f, g). This transient cytokine profile underscores Cu–DHN’s ability to ignite a powerful yet self-limiting immune response. Moreover, no statistically significant differences were observed in hepatorenal function markers, platelet indices, or body weight compared to the PBS control group (Supplementary Fig. 30h). This superior safety was a direct result of our “chemical triggering” design.
Inhibition of distant tumor growth via the ICD effect elicited by Cu–DHN
Pyroptosis, characterized by rapid execution and substantial release of DAMPs and TAAs, prompted us to investigate systemic antitumor immunity in Cu–DHN-treated tumor-bearing mice. Primary orthotopic breast tumors were established through 4T1 cell inoculation in the left mammary fat pad of 6-week-old female BALB/c mice, followed by intratumoral Cu–DHN administration when tumors reached ~200 mm3. Distant orthotopic tumors were induced in the contralateral mammary fat pad 24 h posttreatment (Fig. 6a). CRT labeling of primary tumors revealed significantly increased membrane exposure in Cu–DHN-treated groups (Fig. 6b, Supplementary Fig. 31), indicating effective immunogenic cell death (ICD) and in situ vaccination. Subsequent monitoring demonstrated marked suppression of distant tumor growth in Cu–DHN-treated mice, with complete tumor prevention observed in one subject over 22 days (Fig. 6c, Supplementary Fig. 32). The abscopal antitumor efficacy of Cu–DHN paralleled that of Cu–juglone prototypes, confirming comparable activation of ICD-mediated TAA release and systemic antitumor immunity. In contrast, the Mg–DHN and free CuCl2 + DHN groups showed negligible distant tumor control due to insufficient ICD induction, as evidenced by terminal tumor weight measurements (Fig. 6d). Mechanistic investigations combining flow cytometry and ELISA revealed multimodal immunostimulatory effects of Cu–DHN. Enhanced maturation of DCs in primary tumor-draining lymph nodes (TDLNs) (Fig. 6e, Supplementary Fig. 33), elevated CD8+ T-cell infiltration (although not CD4+ T cells) in distant tumors (Fig. 6f,g, Supplementary Figs. 34, 35), and significantly increased intratumoral TNF-α and IFN-γ levels (Fig. 6h, i). Antibody-mediated T lymphocyte subset depletion experiments established CD8+ T lymphocytes as the principal effectors of systemic immunity (Fig. 6j), with anti-CD8α antibody treatment dramatically abrogating therapeutic efficacy, while CD4+ depletion showed a negligible significant impact (Fig. 6k, Supplementary Fig. 36). These findings collectively demonstrate that intratumoral Cu–DHN administration enables site-specific activation of juglone derivatives, initiates in situ vaccination through ICD-mediated TAA release, promotes DC maturation, and orchestrates CD8+ T-cell-dependent systemic antitumor immunity.

In vivo evaluation of the abscopal effect of Cu–DHN (Cu:DHN:Cys = 0.6:1:0.2). a Schematic illustration of inhibiting distant tumor growth via the abscopal effect of Cu–DHN. b CLSM images of tumor slices labeled with AF488-CRT antibody at 4 days post-treatment. c Longitudinal monitoring of distant tumor growth in BALB/c mice after different treatments (n = 5). d Weights of dissected tumors at 22 days post-treatment (n = 5). e Quantitative matured DCs in primary tumor-draining lymph nodes (TDLNs) (n = 4). The percentage of CD8+ T cells in f CD3+ T cells and g all cells harvested from distant tumors (n = 4). h TNF-α and i IFN-γ contents in distant tumors (n = 4). j Experimental design schematic for investigating CD4+ and CD8+ T cell roles in Cu–DHN-triggered systemic antitumor immunity. k Distant tumor growth curves of 4T1 tumor-bearing mice after Cu–DHN administration along with lymphocyte depletion (n = 5). Data are represented as mean ± SD
Inhibition of pulmonary metastasis via the systemic immune response evoked by Cu–DHN
The highly aggressive pulmonary metastatic propensity of 4T1 mammary carcinoma substantially compromises survival outcomes in tumor-bearing murine models.1 To evaluate the antimetastatic ability of Cu–DHN, orthotopic 4T1 breast cancer models were established in 6-week-old female BALB/c mice through left mammary fat pad inoculation. Upon reaching tumor volumes of ~200 mm3, intratumoral administration of Cu–DHN was performed, followed by intravenous challenge with 4T1 cells 24 h post-treatment to establish pulmonary metastasis (Fig. 7a). Following 14 days of therapeutic intervention, quantitative analysis revealed that Cu–DHN treatment significantly reduced pulmonary metastasis burden, as evidenced by an ~50% decrease in lung wet weight and a >90% reduction in metastatic nodules compared to PBS controls (Fig. 7b–d). This antimetastatic efficacy translated into significant survival prolongation, with Cu–DHN-treated cohorts exhibiting a prolonged survival rate at the experimental endpoint comparable to Cu–juglone prototypes (Fig. 7e).

In vivo evaluation of the anti-metastasis effect of Cu–DHN (Cu:DHN:Cys = 0.6:1:0.2). a Schematic illustration of the mechanism by which Cu–DHN inhibits lung metastasis through systemic antitumor immunity induction. b Lung wet weight and c number of pulmonary metastatic nodules quantified 14 days post-treatment (n = 5). d Representative digital images of lung tissues and histological sections obtained at 14 days post-treatment (the red circles indicate metastatic pulmonary nodules). e Survival rate analysis of 4T1 tumor-bearing mice with pulmonary metastasis following Cu–DHN treatment (n = 8). Quantitative CD8+ T cells in f CD3+ T cells and g all cells obtained from primary tumor-draining lymph nodes (TDLNs) from mice at 3 days post-treatment (n = 3). h TNF-α and (i) IFN-γ contents in serum from mice at 3 days post-treatment (n = 4). j Lung wet weight and metastatic nodule enumeration at 14 days post Cu–DHN administration with concomitant lymphocyte depletion (n = 5). k Survival rate analysis of mice with lung metastasis of 4T1 tumors after Cu–DHN treatment along with lymphocyte depletion (n = 8). Data are represented as mean ± SD
Mechanistic investigations demonstrated the unique capacity of Cu–DHN to convert primary tumors into in situ vaccines, as evidenced by a significant increase in primary tumor-draining lymph node CD8+ T-cell infiltration (Fig. 7f, g, Supplementary Fig. 37) and 5-fold and 3-fold elevation in plasma TNF-α and IFN-γ levels versus the PBS group (Fig. 7h, i). Depletion studies also confirmed CD8+ T cells as pivotal mediators (Fig. 7j, k), with anti-CD8α antibody administration nullifying therapeutic effects (12-fold increase in metastatic nodules), while CD4+ cell depletion showed no significant impact (Supplementary Fig. 38). Notably, Cu–DHN exhibited comparable antimetastatic potency and immunostimulatory effects to Cu–juglone prototypes, attributable to its superior POD activity enabling sustained ROS generation and efficient DHN-to-juglone conversion. This multimodal mechanism synergistically enhanced systemic antitumor immunity, effectively suppressing metastatic progression and improving host survival.
Biosafety of the Cu-DHN prodrug compared with Cu–juglone
The development of pyroptosis-based antitumor therapies faces significant challenges in clinical translation due to the ubiquitous expression of GSDM proteins in normal tissues, which may lead to off-target pyroptosis and subsequent systemic toxicity.4 Herein, we systematically investigated the biosafety profiles of the tumor-specific activatable prodrug Cu–DHN compared with its Cu–juglone prototypes in 4T1 tumor-bearing BALB/c mice (Fig. 8a). Hematological analysis, blood biochemistry, body weight monitoring, and histopathological examination of major organs revealed that Cu–juglone treatment induced severe hepatorenal dysfunction, manifested through abnormal biochemical markers (Fig. 8b–g), characteristic glomerular swelling (Fig. 8j), and thrombocytopenia with reduced plateletcrit (Fig. 8h, i), accompanied by significant body weight loss (Fig. 8k). In contrast, Cu–DHN maintained comparable antitumor efficacy while demonstrating superior biosafety, showing no statistically significant differences in hepatorenal parameters, platelet indices, or histopathological features compared to the PBS group. This safety advantage stems from the tumor-specific activation mechanism of Cu–DHN, where the prodrug remains biologically inert in normal tissues despite partial redistribution from tumor sites (~45% clearance within 4 h post-intratumoral injection, Supplementary Fig. 39). The temporal dissociation between tumor retention and activation kinetics ensures that redistributed Cu–DHN maintains its dormant state in healthy tissues, whereas the constitutive activity of Cu–juglone causes immediate off-target pyroptosis. These findings collectively establish Cu–DHN as a precision pyroptosis inducer that achieves potent local tumor eradication and systemic antitumor immunity activation while circumventing the dose-limiting toxicities associated with conventional pyroptosis-inducing agents, thereby providing a promising platform for developing safe and effective in situ tumor vaccines.

In vivo biocompatibility evaluation of Cu–DHN (Cu:DHN:Cys = 0.6:1:0.2). a Schematic illustration of investigating biosafety of Cu–DHN via biochemical assessment, hematologic analysis, histological sections,and body weight variation. b–g ALT, AST, ALP, BUN, CREA, and LDH contents in serum harvested from 4T1 tumor-bearing mice at 3 days post-treatment (n = 4). h, i Platelets content and thrombocytocrit in whole blood harvested from 4T1 tumor-bearing mice at 3 days post-treatment (n = 4). j H&E staining of histological sections of major tissues at 5 days post-treatment. k Body weight curves of 4T1 tumor-bearing mice during 14 days of Cu-DHN treatment (n = 5). Data are represented as mean ± SD