
Screening and selection
The complete proteome of Acinetobacter baumannii strain ATCC 19,606 (UniProt ID: UP000005476), comprising 3,598 proteins, was retrieved from the UniProt Knowledge Base as the foundational dataset for reverse vaccinology screening. Proteins were analyzed for subcellular localization using multiple prediction tools, and only those consistently identified as extracellular or outer-membrane by at least two independent localization algorithms were retained (Table 1). This stringent overlap criterion yielded 204 proteins, which were considered for subsequent immunogenicity evaluation. Of these 204 proteins, 189 exceeded the antigenicity score threshold, indicating strong potential to elicit an immune response.
In designing the vaccine constructs, the concept of epitope reciprocity was employed to enhance immunogenic potential. This strategy focuses on the strategic placement of B-cell and T-cell epitopes in proximity within the vaccine construct, facilitating cross-epitope interactions. The goal is to intensify immune system recognition by promoting synergistic responses between these two types of epitopes, ultimately leading to stronger and more sustained immune activation.
This prevalence analysis down-selected the list to 15 proteins (Supplementary Data 1 and Supplementary Data 2, and Table 2). Proteins failing any safety criterion were excluded. The remaining 11 proteins, characterized by high antigenicity, extracellular localization, broad genomic occurrence, and absence of allergenic/toxic features, were taken forward as the final vaccine targets. These final candidates are clearly denoted in Table 2 by bold lettering and were subsequently used for epitope mapping and construct design.
Final vaccine candidate selection
Following antigenicity ranking and prevalence analysis, candidate proteins were subjected to safety filtering to remove allergenic or toxic components, ensuring suitability for vaccine design. Allergenicity and toxicity filtering narrowed down the 15 candidates to 11 final proteins (Table 2). These were characterized by high antigenicity scores, extracellular localization, broad genomic presence, and an absence of allergenic or toxic properties, making them optimal vaccine targets. This final panel represents diverse outer-membrane antigens with broad prevalence and safety, providing a robust foundation for epitope mapping. These selected proteins show promise as potential vaccine targets due to their extracellular localization, high antigenicity scores, and frequent occurrence in various strains of A. baumannii. Further experimental validation of these candidates could pave the way for effective vaccine development against this pathogen.
Epitope prediction and selection criteria
Both B-cell and T-cell epitopes were predicted and selected based on explicit scoring thresholds and biological relevance to ensure immunogenicity and safety.
B-cell epitopes
Linear and conformational B-cell epitopes were successfully identified and prioritized based on high predictive scores. The top-ranked epitopes were retained for construct integration (Supplementary Data 3).
T-cell epitopes
T-cell epitopes were identified to ensure coverage of both cytotoxic (MHC I-restricted) and helper (MHC II-restricted) immune responses. MHC I and MHC II binding peptides were predicted using NetMHCpan-4.1 and NetMHCIIpan-4.3, respectively. MHC I epitopes were retained as strong binders with percentile rank ≤ 0.5%, and MHC II epitopes with percentile rank ≤ 2%. Cytotoxic T-lymphocyte (CTL) epitopes were predicted with NetCTLpan-1.1, applying proteasomal cleavage score ≥ 0.8, TAP transport score ≥ 0.5, and MHC binding percentile ≤ 0.5%.
Conservation and safety filtering
All epitopes were assessed for sequence conservation across A. baumannii genomes, retaining those with ≥ 85% identity in homologous sequences. Potential cross-reactivity was evaluated by BLASTP against the human proteome, excluding epitopes showing > 35% identity over ≥ 9 consecutive residues.
This filtered set of epitopes formed the building blocks for subsequent multi-epitope construct design and adjuvant integration (Supplementary Data 3). The selected B-cell epitopes offer structural compatibility with T-cell epitopes for integration into multiepitope constructs.
Adjuvant selection
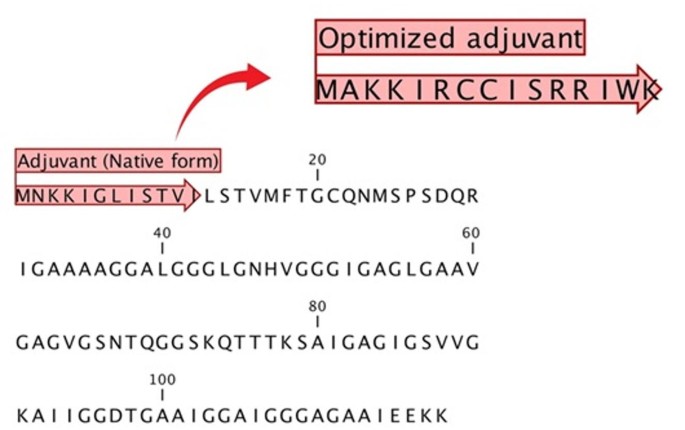
An immunomodulatory adjuvant sequence was selected to enhance vaccine potency by promoting immune activation. The YMGG-like Gly-zipper domain-containing protein, identified with the accession number D0CB14 and an antigenicity score of 1.173, was selected as the adjuvant for this study. The sequence of this protein is illustrated in Fig. 1. The selection of this protein was based on its high antigenicity, which exceeded the established threshold, and the presence of IgA epitopes. This adjuvant’s high antigenicity and IgA epitope content predict strong mucosal immune response potential.

Sequence details of the selected adjuvant. The whole sequence of the adjuvant is presented, with key regions marked by red arrows. The upper right section illustrates the optimized adjuvant sequence, highlighting modifications introduced for enhanced immunogenic properties.
Table 3 presents the N-terminal segment of the protein, predicted as an immunomodulatory region, along with its corresponding scores.
After undergoing site-directed mutations, the sequence was optimized, resulting in the following engineered sequence. The properties of this modified sequence are shown in Table 4.
Adjuvant design
The adjuvant peptide integrated into the final multi-epitope construct (Figs. 1 and 2; Tables 3 and 4) was derived from an A. baumannii protein harboring a YMGG-like Gly-zipper domain (UniProt ID: D0CB14). The native fragment exhibited strong predicted antigenicity (VaxiJen v2.0 score = 1.173) and included putative IgA-stimulatory epitopes. The most reactive 11-mer identified using VaxinPAD was optimized by site-directed peptide engineering, modifying residue composition to improve charge balance and hydrophobic stability while maintaining TLR-binding capacity. The optimization yielded a 15-mer peptide with increased predicted antigenicity (0.86 vs. 0.15 for the native peptide) and immunomodulatory SVM scores > 3.0.

Contact map and structural representation of the designed construct. The upper left panel displays the contact map score for the construct, with the amino acid sequence represented along the x- and y-axes. The color gradient ranges from blue (low score) to red (high score), with a red box indicating the probable interface region between two identical construct sequences. Green dots represent experimentally confirmed contacts extracted from the PDB database. The upper right section shows the complete amino acid sequence of the construct, with predicted interface residues highlighted in red, corresponding to the red box in the contact map. The lower panel provides a schematic illustration of the homotrimer assembly, demonstrating the integration of three identical subunits.
Docking simulations were conducted against TLR-4, TLR-1, and TLR-3 to assess broad receptor interaction potential. TLR-4 was included as the canonical receptor for bacterial immunomodulatory motifs; TLR-1 docking verified compatibility of the adjuvant surface within the vaccine construct; and TLR-3 docking evaluated endosomal receptor engagement. These analyses collectively indicated favorable binding affinities across receptor types, validating the peptide’s suitability as an optimized adjuvant sequence with multi-TLR activation capacity.
Structural design
To provide a context and rationale for the vaccine construct design, the overall approach combined immunological, structural, and physicochemical principles. The multiepitope construct was arranged to integrate B-cell and T-cell epitopes in reciprocal proximity, maintaining antigenic diversity while preserving structural compactness. This configuration was guided by predicted antigenicity (VaxiJen v2.0 scores ≥ 0.4), safety filtering (AlgPred2, ToxinPred2), and linker selection rules described earlier, ensuring optimal spatial presentation of immunologically active sites. The adjuvant was positioned at the N-terminus with an EAAK rigid linker to stabilize orientation and distinct domain separation. Construct length and composition were balanced to maintain molecular weight within 22–26 kDa and isoelectric pH near 9, parameters compatible with both structural feasibility and antigen accessibility (Fig. 3). These design decisions collectively ensured that the model possessed a plausible quaternary structure and predicted capacity to multimerize, justifying subsequent analyses by MultiFOLD and GalaxyHomomer for higher-order stability evaluation.
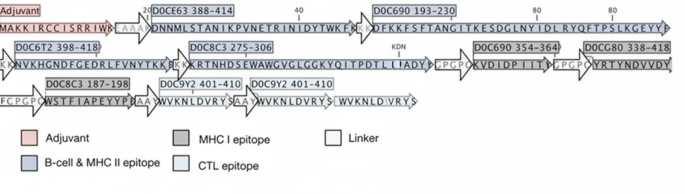
Detailed structural composition of the designed construct. Each region of the construct is labeled with specific tags, as indicated in the image. The color key at the bottom corresponds to each tagged region, providing a clear visual guide to the distinct sections within the construct’s sequence.
Following the selection of a set of epitopes, four constructs were designed, and their three-dimensional structures were predicted using multiple software tools. To maximize cooperative immune activation, the constructs were organized according to the principle of epitope reciprocity, which involves spatially associating B-cell and T-cell epitopes to promote cross-epitope signaling. Flexible linkers were strategically applied to preserve epitope conformation while positioning complementary epitopes in close proximity. This arrangement was predicted to enhance immune recognition and synergistic activation.
Each model was assessed through structural validation and physicochemical analyses, including MolProbity scoring, RMSD comparison, stability index, and VaxiJen v2.0 antigenicity values (Table 5). The construct demonstrated the highest structural quality and antigenicity, along with an instability index below 40, was selected as the best model and used for subsequent analyses. Figure 3 shows the details of this optimized construct.
To explicitly assess trimerization capacity, the designed construct was analyzed using in silico quaternary-structure prediction platforms. The amino acid sequence was first submitted to MultiFOLD, which integrates consensus threading and ab initio modeling to evaluate oligomeric states. MultiFOLD predicted a strong tendency for homotrimer formation with favorable interface scores and high global model quality estimates. To independently validate this prediction, the monomeric structure refined in UCSF ChimeraX was subsequently processed through GalaxyHomomer, a template-based modeling server specialized for oligomeric assembly assessment. GalaxyHomomer consistently returned a stable homotrimer configuration, characterized by cohesive inter-chain contacts and minimal steric conflict. Together, these findings confirmed that the construct was structurally capable of adopting trimeric organization while maintaining the architectural integrity required for effective epitope presentation.
The results obtained from the MultiFold software suggested the possibility of multimeric behavior in the constructs. Upon providing the construct sequence, MultiFold predicted a homotrimer configuration, a protein structure comprising three identical polypeptide chains (Fig. 4).

The trimeric configuration of the designed construct is shown from three different perspectives (a-c), each highlighting structural organization and spatial arrangement. The color key beneath the figures (d) provides a schematic of the construct, with color harmonies consistently applied across all sections, representing functional regions accurately and uniformly.
To determine how the principle of epitope reciprocity influenced construct architecture, reciprocal epitope mapping was performed using the refined structural model. The final multiepitope design ensured that predicted B-cell epitopes were spatially adjacent to corresponding helper T-cell epitopes, fostering potential cross-communication during immune recognition. Structural visualization in UCSF ChimeraX revealed that reciprocity-based placement reduced steric hindrance and maximized surface exposure of paired epitopes, confirming their mutual accessibility. To evaluate the functional relevance of this design, additional comparative models were generated without reciprocity constraints. In these alternative arrangements, several key epitope pairs showed disrupted orientation and lower solvent-accessible surface areas, suggesting suboptimal antigen presentation. Quantitative interface analysis from GalaxyHomomer and MultiFOLD confirmed that reciprocity-optimized constructs displayed greater structural coherence and higher stability indices than non-reciprocal counterparts. These findings emphasize that epitope reciprocity is not only a conceptual design feature but also a structural determinant of epitope accessibility and overall vaccine architecture integrity.
To further optimize the structure and confirm the homotrimeric nature of the construct, the predicted monomers from MultiFold were isolated and refined using the UCSF ChimeraX software. The refined structures were then submitted to GalaxyHomomer for fourth structure prediction, with the software automatically determining the multimeric state. Once again, a homotrimer was predicted, reinforcing the likelihood that the designed construct adopts a homotrimeric form.
To assess whether the designed antigen could adopt a higher-order multimeric form while retaining monomeric integrity, we employed in silico quaternary-structure prediction tools. First, the amino acid sequence of the construct was analyzed using the MultiFOLD server, which predicts tertiary and quaternary structures by integrating multiple threading and ab initio models. MultiFOLD identified a strong propensity for homotrimer formation, evidenced by high interface scores and favorable global model quality estimates. To independently verify this prediction and optimize structural stability, the monomeric model generated was refined in UCSF ChimeraX and submitted to GalaxyHomomer, a template-based modelling platform specialized for oligomeric assembly prediction. GalaxyHomomer consistently returned a homotrimer configuration with stable inter-chain interfaces, supporting the likelihood that the designed construct would adopt this form under physiological conditions. Both analyses demonstrated that potential multimerization did not compromise predicted antigenicity or epitope accessibility, thereby supporting the inclusion of multimerization capacity as part of the construct design rationale.
Table 5 presents the properties of four vaccine constructs, evaluating various attributes such as stability, antigenicity, and molecular weight. Among the four designed constructs, comparative evaluation revealed that all candidates met baseline safety criteria (non-allergenic, non-toxic) and stability indices below 40. However, the construct 2 demonstrated the optimal overall profile, with the highest antigenicity score (0.80), ideal molecular weight (24.09 kDa), and moderate isoelectric point (pH 9.28). Its predicted three-dimensional organization showed compact folding and consistent trimeric assembly as suggested by MultiFOLD and GalaxyHomomer analyses. These features collectively supported selection of the construct 2 as the final vaccine model illustrated in Fig. 3, and Fig. 4. Table 5 retains comparative data for all four constructs to maintain transparency in the development and evaluation process.
Structural and functional validation
The finalized constructs demonstrated favorable properties, including stability indices below 40, molecular weights ranging from 22.57 to 25.72 kDa, and high antigenicity scores. No allergenic or toxic epitopes were detected in the constructs.
This streamlined pipeline from data acquisition to final construct design highlights the immunogenic potential of these targets, laying the groundwork for future experimental validation and potential clinical application as a multiepitope vaccine against A. baumannii.
Docking results
To ensure comprehensive receptor profiling, molecular docking simulations were performed against TLR-4, TLR-1, and TLR-3 using distinct computational tools optimized for each receptor type. The HADDOCK platform (Supplementary Data 1) was applied to TLR-3 due to its endosomal localization, while TLR-1 docking was performed with LZerDock to assess surface receptor engagement and adjuvant orientation. A parallel TLR-4 docking run served as an additional verification of canonical bacterial pattern–recognition motif compatibility. These combined analyses provided multi-receptor validation of the construct’s interaction potential and ensured consistency between the main text and supplementary materials.
To determine whether the engineered construct possessed attributes suitable for further preclinical evaluation, we conducted a comprehensive post-design analysis. This included structural mapping of conformational epitopes and simulation of receptor interactions to judge the likelihood of triggering effective immune responses.
The docking study with LZerDock generated several models, with the best configuration assigned for the model presented in Fig. 5. This configuration was notable for its alignment of the construct’s adjuvant domain toward the TLR-1 receptor, supporting the expected interaction hypothesis. The scoring results are detailed below:

The complex of the designed construct with Toll-like receptor 1 (TLR1). The correct orientation of the construct toward TLR1 is depicted, with the construct’s adjuvant region highlighted in red and the TLR1 receptor in dark blue. The construct is shown in a light blue-ribbon representation with a 70% transparent surface overlay, illustrating the spatial interaction and docking alignment between the construct and TLR1.
GOAP score
−106742.06, ranked 223 among all models.
DFIRE score
−93186.25, ranked 132.
ITScore
−45072.09, ranked 125.
Ranksum score
480.
These scores collectively indicate a favorable binding interaction between the construct and TLR-1. The negative GOAP and DFIRE values suggest a stable conformation, while the relatively high ITScore aligns with a strong binding interaction at the molecular level. The rank-sum score confirms the model as one of the best-fit configurations across all docking parameters.
The docking analysis via Mapiya revealed a significant interaction between the adjuvant segment of the vaccine construct and TLR residues. The contact map highlighted several critical interface residues. Notably, key residues from the adjuvant components of the construct were in proximity to residues in the TLR receptor, indicating a strong interaction (Fig. 2). These interactions were consistent with the intended immunogenicity design, which aimed to ensure the adjuvant’s proximity to TLRs for optimal immune activation.
Contact map findings
The contact map demonstrated that specific adjuvant residues form close contact with TLR residues. These predicted interactions from the in silico docking reinforce the anticipated role of the adjuvant in potentially triggering immune responses (Fig. 6), noting that these findings are computational predictions without experimental validation.

Contact map of the construct and TLR1 complex. The interactions between the adjuvant residues of the construct and TLR1 are highlighted in green, showing clear and significant contacts. This map provides a detailed view of the interface and specific points of contact between the adjuvant region of the construct and TLR1, confirming the intended binding orientation.
The molecular complex, visualized through a 3D image and analyzed via Mapiya, further supported the construct’s docking accuracy. The close alignment of these interacting residues emphasizes the effectiveness of the designed construct in interfacing with TLRs, which is essential for inducing a potent immune response. Figures 7 and 8 present the computationally simulated immune response following three hypothetical vaccine administrations, as predicted by the C-IMMSIM platform.

The graph representation of antibody titer and antigen count simulated for three virtual injections of the vaccine. X and Y axes are labeled, and the color key for defining the curves is presented in the image.

The graph represents the count of the active B-cell population.