

HSP60 peptides as in vitro inducers of the TLR4/MD-2 complex
In order to determine the potential effect of HSP60 peptides of M. bovis on the TLR4/MD-2 complex, the activation capacity was evaluated by measuring cell viability and the overexpression of two key proteins in the signaling pathway: The TLR4 receptor and IL-6. This exploration was carried out in human and mouse endothelial cell lines, HCAEC and C166, respectively. First, the effect of peptides 1–6 on human cell viability was determined. Figure 2A shows that incubation of the peptides and LPS for 24 h did not induce a significant decrease in cell viability. Previous studies have shown that concentrations between 100 and 1000 ng/mL of LPS do not alter cell viability nor the expression of certain cytokines, such as TNF-α, but do induce the overexpression of direct activation markers of the TLR4/MD-2 complex, such as IL-647. IL-6 is the cytokine that exhibits the highest degree of overexpression in response to TLR4 activation by various molecules, such as LPS. IL-6 has been shown to play a major role in endothelial cell dysfunction during acute inflammatory events in microvascular, macrovascular, bladder, and umbilical vein endothelial cells, and its presence also induces an increase in endothelial cell permeability48,49.
To determine whether this treatment was able to induce the overexpression of IL-6, the presence of this mature and exocytosed cytokine was measured in the supernatants of the treated cells. In Fig. 2B, it can be observed that incubation with LPS for 24 h was able to increase the presence of IL-6 by around 5.5-fold with respect to the control group; however, none of the peptides had an effect on the receptor, as no increase in the presence of IL-6 was shown. Due to the inability of peptides to activate the TLR4/MD-2 complex and in order to determine whether the increase in IL-6 in the LPS-treated group was directly mediated by TLR4 activation, cells were incubated with the inhibitor TAK-242, which is specific for inhibiting TLR4 receptor signaling. Figure 1B and Supplementary Fig. 1 A (S1A) and SB show that cell viability is not altered when experimental treatments (LPS, P1-6, and BSA) were pre- and co-incubated with the TAK-242 inhibitor and that LPS-mediated IL-6 overexpression was prevented by pre-incubating cells with this specific inhibitor. This allowed us to determine that HCAECs are capable of being activated by LPS, the classic agonist of the TLR4/MD2 complex, but that the peptides did not induce a biological effect under these conditions.
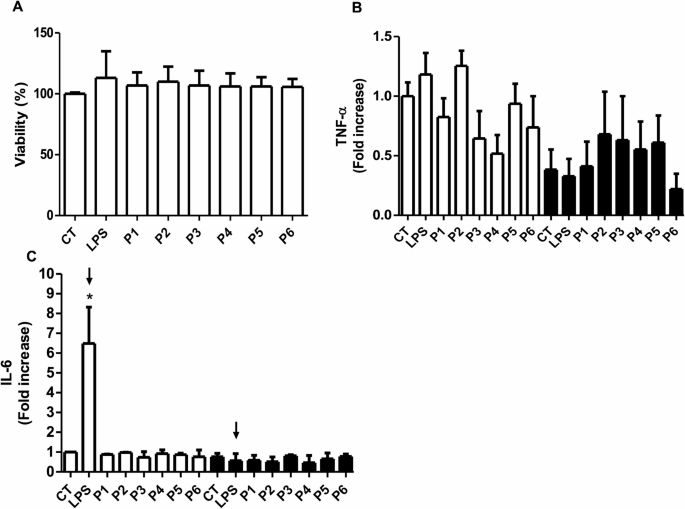
Effect of the treatment with HSP60 peptides and lipopolysaccharide on endothelial cells (HCAEC). A) Viability of endothelial cells after 24 h of treatment with peptides (P) (100 ng/mL) and lipopolysaccharide (LPS) (100 ng/mL). B and C) Tumor necrosis factor-alfa (TNF-α) and Interleukin 6 (IL-6) expression following treatment with HSP60 peptides and LPS (white bars) and by coincubation with 1 µM TAK-242 (black bars). Arrows mark the LPS-treated group, with and without the TLR4 inhibitor. *p < 0.001 vs. control (CT). n = 4–5.
In order to compare the results obtained in human endothelial cells, an exploration of the effect of the peptides and LPS on cell viability and activation of the TLR4/MD-2 complex in mouse endothelial cells was carried out. To determine the potential effect on cell viability mediated by LPS, a concentration-increasing curve was performed to determine the effect of this molecule. Figure 3A shows that LPS was capable of mediating a dose-dependent decrease in cell viability, which allows us to continue determining the potential effect of peptides on this cell model. To confirm that LPS is capable of increasing TLR4 production and thus IL-6 overexpression, the protein expression level of these two proteins was measured. Figure 3B shows that LPS treatment is capable of significantly increasing TLR4 expression compared to the control group, results that are consistent with those reported in the literature7,8. In addition, in order to determine that this overexpression was related to the production of one of its products, we measured IL-6 levels. Figure 3C shows that LPS treatment induced a significant increase in this cytokine compared to the control group, showing results consistent with those reported in the literature12,48. Complementary images can be found in Figure S11.
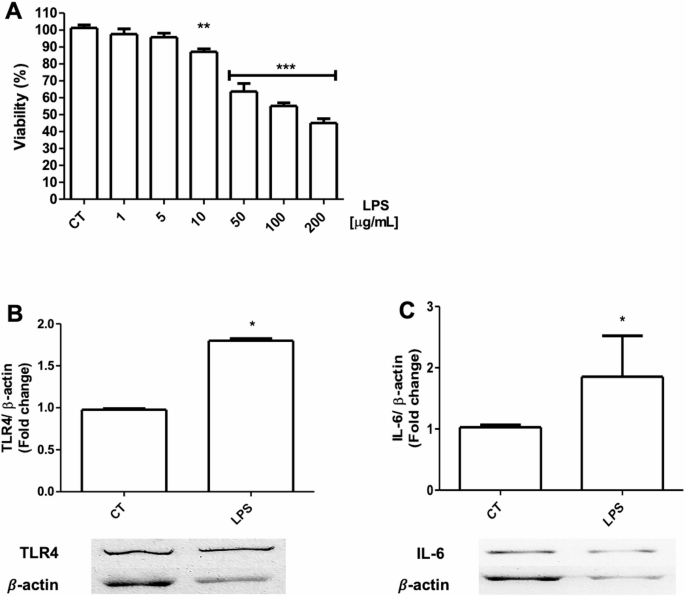
Dose-response curve with lipopolysaccharide at different concentrations and expression of protein activation markers on endothelial cells (C166). A) Viability of endothelial cells after lipopolysaccharide (LPS) treatment at different doses (1–200 µg/mL). B) Expression of Toll-like receptor 4 (TLR4) on endothelial cells treated with LPS (100 µg/mL). C) Expression of Interleukin 6 (IL-6) on endothelial cells treated with LPS. *p < 0.05 vs. control (CT), n = 4–5.
Once our cell model was validated, we proceeded to determine the potential effect of peptides 1–6 on cell viability in C166 cells. In Fig. 4A, it can be observed that incubation with peptides 1, 2, 3, 5, and 6 (P1, 2, 3, 5, and 6) did not induce a decrease in cell viability; however, P4 showed a significant decrease with respect to the control group. Given this significant difference between P4 and the other peptides, we explored whether P4 could mediate a dose-dependent decrease in cell viability. Figure 4B shows that cell viability decreases in a dose-dependent manner. Once the experimental conditions for modulating cell viability with LPS and peptides were established, it was proceeded to determine whether these treatments were capable of modulating TLR4 and IL-6 expression. Figure 4C shows that P4 and P5 are capable of significantly inducing TLR4 overexpression, unlike P6, which maintains levels similar to the control group. However, TLR4 overexpression only induced a significant increase in IL-6 mediated by P4, not showing a significant effect with treatment with P5 and P6 (Fig. 4D). Complementary images can be found in Figure S11.
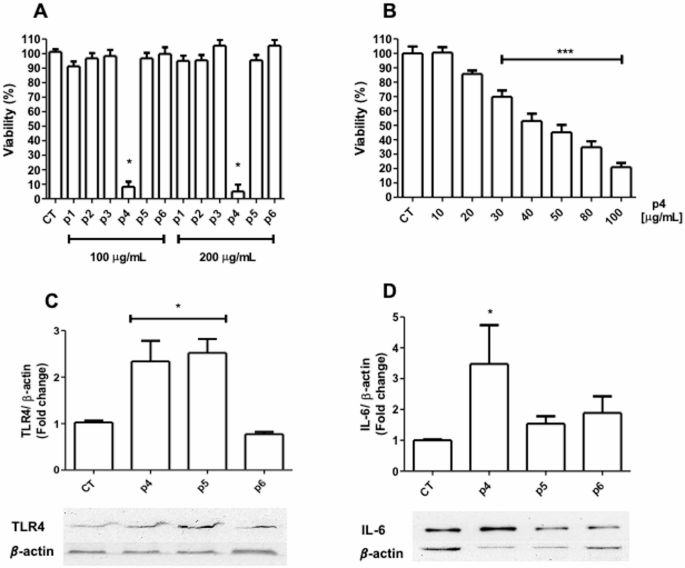
Effect of the treatment with HSP60 peptides on mouse endothelial cells (C166). A) Viability of endothelial cells after peptides (P1-6) treatment at different doses (100 and 200 µg/mL) for 72 h. B) Dose-response curve with peptide 4 (P4) at different concentrations (10–100 µg/mL) after a 24 h treatment. C & D) Expression of Toll-like receptor 4 (TLR4) and Interleukin 6 (IL-6) on endothelial cells treated with P4, P5, and P6 at 40 µg/mL for 24 h. *p < 0.05 vs. control (CT), n = 3–4.
To determine whether the coupling, adaptation, and potential interaction of the peptides with the TLR4/MD-2 complex, due to its size, is capable of inducing IL-6 production or mediating a direct effect on cell viability, cells were incubated a protein different from HSP60, total albumin (BSAc) and BSA digested (BSAd) with proteinase K, producing various peptide fragments of around 3 to 18 amino acid residues. As observed in Figure S1A and SB, cells with and without TAK-242 inhibitor showed an alteration in cell viability in the presence of BSAc and BSAd. On the other hand, neither BSAc nor BSAd were able to produce an increase in the production and exocytosis of IL-6 compared to the group treated with LPS (Figure S1B), showing that not any random sequence of amino acid residues that docks in the TLR4/MD-2 complex can activate this complex.
Molecular docking of the peptides and LPS with the dimeric TLR4/MD-2 complex
This analysis aims to explore molecular interactions that may explain the observed peptide effects, in particular P4 and P5, responsible for IL-5 and TLR4 expression in mouse cells, respectively. Recently, positive effects in the use of TLR4 modulating peptides in macrophages and mouse models of inflammatory bowel disease have been reported, indicating the activation of TLR4, nonetheless, there is no proposal of the peptide-receptor binding mode at the molecular level nor crystallographic structures of the resulting complex50. Molecular docking assays were performed to decipher potential differences in the binding site of the TLR4/MD-2 complex. Numerical results from the docking simulations are presented in Table 1. For LPS and its derivatives, the docking results correspond to the pose most closely aligned with the reference crystallographic structure; therefore, a single value is reported for both the free energy of binding (ΔGbinding) and the root mean square deviation (RMSD, Table 1).
We analyzed the ten highest-energy peptide poses from VINA docking. Because peptides can engage receptors via multiple binding modes and no crystallographic evidence dictates a single interaction pattern, all docking poses were considered to capture the full range of potential contacts. This exploratory approach, while not identifying the sole biologically active pose, highlights the most robust interactions and the chemical features that orient peptides within the binding site. The conformational plasticity of the peptides and absence of reference structures motivate an inclusive analysis to identify residues critical to peptide–receptor interactions and to quantify their contributions. Table 1 presents both ranges and average values.
We explored the theoretical ΔGbinding of LPS derivatives and peptides. Lipid A (LA) showed the lowest binding energy (highest affinity) in both species, reflecting high ligand-receptor complementarity and predominant van der Waals contributions. The Ra-LPS chemotype (PDB 3FXI), consisting of LA and a core oligosaccharide lacking the O-antigen3, exhibited binding energies comparable to those of peptides, although its large size and high flexibility limit docking accuracy51,52.
Peptide binding energies ranged narrowly from − 5.76 to − 7.75 kcal/mol, corresponding to roughly a 100-fold variation. Peptides P2 and P3 displayed the lowest ΔGbinding values for the mouse receptor but were biologically inactive, whereas P4 and P5 (− 6.60 Kcal/mol, −6.35 Kcal/mol, respectively), despite weaker binding energies, induced TLR4 and IL-6 expression.
These results indicate that ΔGbinding alone is not a reliable predictor of biological activity, consistent with reports showing that the highest-affinity pose does not necessarily correspond to the active one53,54. Moreover, analysis of peptide-receptor contacts revealed that a higher number of interactions does not imply stronger binding, as decoy peptides with extensive contacts still exhibited moderate binding energies, confirming that interaction count alone is also not predictive of activity (Table S1). Similarly, the ΔGbinding calculations performed with PRODIGY reveal a higher affinity of peptide P6 for the mouse TLR4 receptor complex compared to P4 and P5, further supporting the lack of correlation between the predicted binding energy and the observed experimental effect (Table S5).
Although P4 and P5 exhibit comparable binding energies to Ra-LPS, their distinct biological responses suggest that these peptides may engage and activate the TLR4/MD-2 complex through mechanisms different from those of canonical LPS-derived ligands53,54.
These observations indicate that activation of the TLR4/MD-2 receptor complex depends on specific structural and chemical requirements that enable dimerization and subsequent activation, rather than being dictated solely by isolated parameters such as ΔGbinding or the total number of interactions formed by a peptide. Instead, activation is determined by both the quantity and the nature of specific interactions, which in turn arise from the amino acid composition of the interacting peptide53,54.
Peptide flexibility does not necessarily confer an advantage, as biologically active sequences tend to adopt conformationally consistent binding modes that favor specific, functionally relevant contacts. This observation, consistent with reports for the activating peptide Neoseptin-3, underscores that conformational stability rather than binding energy per se better predicts the potential for TLR4/MD-2 activation55.
On the lack of a molecular description of the modulatory mechanism, in silico approaches have provided the theoretical insight into the molecular interactions, showing that peptides are prone to cluster into specific TLR4 conserved regions with a vast pose dispersion, showing that residues in interaction rather than binding affinity are relevant in the definition of novel modulators12.
Interaction frequency of HSP60 peptides: the case of P4, P5, and P6 on the dimeric complex TLR4/MD-2
Regarding the RMSD, this value indicates the average conformational variation among the ten poses generated by docking concerning the initial pose; therefore, the larger the value, the greater the variability in conformations that can be accommodated within the binding site. In terms of biological activity, however, it does not provide relevant information in this case. As free energy binding values and RMSD failed to account for the presence or absence of biological activity, an extensive and detailed computational analysis of the binding site interactions was conducted for each pose of every peptide, aiming to discover molecular-level insights. This analysis was conducted using Python (or PyMOL) molecular interaction analysis libraries with the script described in the methods section.
Since P1, P2, and P3 showed no effect in either species, we focused on P4, P5, and P6 as a case study (Fig. 5) related to their in vitro activity in both human and murine cells. It is important to recall that P4 induced an increase in TLR4 and IL-6 expression, P5 increased TLR4 expression, and P6 did not exhibit biological activity but displayed a particular interaction profile with a high binding energy. The interaction frequency of each peptide residue across all poses is shown in Fig. 5. This approach allows for the identification of peptide residues with high interaction potential, particularly those likely to play a critical role in the biologically active peptides (green box, Fig. 5).

Bar plots of the frequency or number of interactions (x-axis) between each residue (y-axis) of HSP60 P4, P5, P6, and the human (h) and mouse (m) dimeric TLR4/MD-2/TLR4* receptor complex. The horizontal black lines are placed every five residues as reference. Peptides exhibiting in vitro activity in this work, P4 and P5, are highlighted in green.
Interaction frequency plots reveal that P4 and P5 engage the mouse TLR4/MD-2 receptor mainly through their N-terminal regions, where non-contiguous acidic residues form high-frequency contact clusters. These interactions correlate with biological activity, while peptides lacking activity (P1, P2, P3, P6) display uniformly low or C-terminal interactions, often involving fewer than fifteen contacts (Fig. 5). The results for P1, P2 and P3 are presented in the supplementary material section (Tables S1, S2, S3, and S4, and Figures S2 and S3).
Although P6 shows an extensive C-terminal interaction profile, its inactivity indicates that neither the total number of contacts nor local acidity in this region is sufficient for activation. Instead, the alternate or non-contiguous distribution of negative charges near the N-terminus appears crucial for receptor engagement. Comparative alignment of all peptides (Fig. 6) supports this observation, highlighting that only those with strategically positioned acidic residues at the N-terminal region effectively promote TLR4/MD-2 mediated effects.

Multiple Sequence Alignment-Peptides. Position-based sequence alignment of all 15-amino acid peptides evaluated in vitro and by docking. Blue: positively charged residues, red: negatively charged residues, magenta: aromatic residues, gray: neutral hydrophilic residues.
In the human model, P4 and P5 also display N-terminal interaction predominance, though at lower frequencies than in mice. This supports that, in mouse TLR4, the presence of at least three non-contiguous acidic residues near the N-terminus favors activation, whereas this feature alone does not elicit activity in humans (Figs. 5 and 6).
Docking simulations with decoy peptides (15Ala, 15Arg, 15Glu, 15Gln, 15Asp) revealed that charge and hydrogen bonding capacity jointly influence receptor interaction. Acidic and polar decoys (15Gln, 15Asp, 15Glu) showed uniformly high interaction frequencies and contacts with chain B residues, exceeding the interactions exhibited by active peptides (Figures S4–S7). Also BSA-derived peptides, despite high interaction frequencies (notably Glu2 and His6), failed to activate TLR4/MD-2, consistent with prior evidence of BSA’s non-agonistic or even anti-inflammatory behavior in endothelial cells56,57,58.These findings highlight that effective activation of the TLR4/MD-2 complex requires not only canonical interactions but also precise, functionally relevant interactions (Figure S8 and Figure S9).
In mouse models, mAsp5(P4) and mGlu2(P5) exhibited the highest interaction frequencies, correlating with receptor activation. These residues are both negatively charged and located in the N-terminal region, reinforcing the functional relevance of these residues in this segment. Previous studies have reported the importance of acidic residues in peptide-mediated activation of the TLR4 receptor. In particular, mutation of Asp2 with alanine (Ala) in the SPA4 peptide significantly reduced its activity. Similar findings have been observed in long protein sequences derived from HMGB1, underscoring the critical role of acidic residues in promoting receptor activation59,60.
Altogether, these results suggest that TLR4 activation depends on both the conformational arrangement of acidic residues and the peptide orientation at the binding site. This insight supports the rational design of shorter analogs (5–10 residues) targeting key N-terminal motifs while accounting for multi-targeting challenges.
This analysis enables the identification, within each peptide, of the residues or regions that are critical for establishing interactions that drive receptor modulation7. The higher the number of interactions involving a given residue, the greater the number of docking resulting conformations that exhibit that interaction61. It can be inferred that peptides lacking biological activity interact with the receptor complex in a less specific way. In contrast, P5 and especially P4 are capable of forming alternating, well-defined point interactions characterized by favorable orientation, directionality, and propensity. While these interactions may not be strictly specific or fully complementary, they appear to play a key role, supporting the peptides’ capacity to activate the receptor.
Interaction frequency profiles of the dimeric TLR4/MD-2 residues with HSP60-derived peptides
Shifting the focus toward the receptor, the residues of the dimeric TLR4/MD-2 receptor complex involved in interactions with the ten poses of each peptide were identified, and the frequency of interactions established by each residue was quantified and plotted (Fig. 7).
In the human receptor complex, peptides show limited interaction with chain B (TLR4*), correlating with the absence of biological activity. In contrast, murine models exhibit a higher density of contacts, particularly involving residues mSer413(B) and mArg434(B), which appear recurrent across active peptides (Table 2). However, frequent contacts with these residues alone do not ensure activation, as illustrated by P6, which, despite a comparable number of interactions, remains inactive.

Bar plots of the frequency or number of interactions (y-axis) between HSP60 peptides P4, P5, P6, and each chain residue (x-axis) of the human (h) and mouse (m) dimeric TLR4/MD-2/TLR4* receptor complex (A/C/B chains). The red lines are to differentiate between the different chains of the receptor complex, from left to right: First third: Chain A (TLR4), second third: Chain B (TLR4* or counter TLR4), and third third: Chain C (MD-2). The y-axis scale is the same in all graphs. This multi-conformational analysis was performed exclusively with peptides; therefore, only peptide-related plots are shown. Peptides exhibiting in vitro activity in this work, P4 and P5, are highlighted in green.
Active peptides P4 and P5 exhibit interactions with key residues on both TLR4* and MD-2 (chains B and C, Table 2; Fig. 7), suggesting that functional activation depends on specific residue identity and distribution rather than overall contact number. In particular, the non-contiguous arrangement of acidic residues in the N-terminal region appears to be decisive for mouse receptor activation, a feature absent in humans. Although P2 has previously been reported to exhibit activity in rat cells2, it showed no biological effect in either human or mouse models in our study. Thus, the functional relevance of acidic residue distribution in peptide-mediated TLR4 activation appears to be related to the murine receptor2.
Unlike LPS, whose acyl chains engage MD-2 via Phe126(C) to promote dimerization, peptide binding likely induces activation through a more direct mechanism, facilitating chain B contact without accessory proteins (CD14 or LBP). This implies that peptide-induced dimers may differ structurally from those generated by LPS62,63,64,65.
These results highlight TLR4* as the critical interface for activation and underscore that successful signaling requires a precise spatial configuration of acidic residues and contact points and is not related to the total number of interactions.
Table 2 reveals that activation of the human TLR4/MD-2 complex demands highly specific interaction patterns, as none of the tested peptides reproduced the precise contacts observed for LPS. Although all peptides interact with hSer415(B), this residue does not appear to be essential for activation. In mice, the opposite occurs: fewer yet strategically positioned interactions seem to facilitate receptor activation. The homologous residue mSer413(B) is contacted by nearly all peptides but does not, on its own, account for biological activity.
Among the tested sequences, P4 and P5 stand out for establishing discrete, functionally relevant interactions rather than maximizing contact number or binding energy. In particular, mAsp393(B) and mMet412(B) form key contacts with Met4(P4), Asp5(P4), and Glu2(P5), defining the minimal interaction pattern associated with receptor activation. These residues delineate a segment within chain B whose engagement appears critical for triggering TLR4 expression and dimer stabilization.
The arrangement and spacing of acidic residues in P4 and P5 enable a coherent network of alternating interactions along the activation region (mSer386–mSer439)7,19. P4 uniquely engages mAsp393(B), correlating with its stronger biological response, whereas P5 reproduces the same pattern only partially. In contrast, P6 forms numerous yet functionally irrelevant contacts, underscoring that activation depends on the precision and diversity of interactions rather than on their abundance.
The molecular interactions formed by LPS and the ten docking poses of peptides P4, P5, and P6 within the binding site of the dimeric TLR4/MD-2/TLR4* receptor complex (A/C/B chains) in both human and murine species are illustrated in Fig. 8. In mice, LPS establishes only two interactions with chain B (TLR4*), one electrostatic and one π-stacking, whereas in humans, it forms nine interactions, including hydrogen bonds and π-stacking. This suggests a restrictive activation mechanism in mice, with fewer and less diverse interactions, underscoring the critical role of the binding region, whereas in humans, a permissive activation mechanism by the TLR4/MD-2 complex as an evolutionary defense mechanism.

Molecular interactions identified for LPS and the ten docking-generated poses of peptides P4, P5, and P6 interacting with the mouse (m) dimeric TLR4/MD-2/TLR4* receptor complex (A/C/B chains). LPS is shown as a cyan stick representation. For the peptides, all ten docking poses are superimposed and displayed in translucent cyan, with the highest-energy pose highlighted in a more defined stick format. Interaction analysis was performed using a custom script based on PyMOL libraries, and molecular images were generated using PyMOL Viewer52. Interacting residues from receptor chain A (TLR4) are highlighted in blue, chain B (TLR4) in orange, and chain C (MD-2) in light yellow. Residues mentioned in the text are annotated with their corresponding chain letter in parentheses following the residue number. For example, the residue Met412 highlighted in orange in the figure is referred to as Met412(B) in the text. Interactions color code: Red: Electrostatic, Blue: H-bond, White: π-cation/anion, Green: Salt-Bridge. The images of the same ligands interacting with the human TLR4/MD-2/TLR4* receptor complex are shown in the supplementary material (Figure S10).
The binding mode of each peptide is determined by its sequence. Although peptides may adopt multiple conformations, those associated with biological activity consistently engage specific residues through defined orientations. Thus, peptides with a higher propensity to adopt such conformations, favoring interactions with key activation residues, exhibit greater potential for showing biological effects (Fig. 8)2.
To further validate the functional relevance of specific residues, mutagenesis studies of P4 and P5 are currently being conducted both in vitro and in silico. These experiments seek to confirm whether the peptide residues previously associated with activation, particularly Met4 and Asp5 in P4 and Glu2 in P5, are indeed responsible for the biological effects observed. Moreover, residues such as Glu2 in P4 and Asp6 in P5, which appear to act primarily as anchoring points rather than activation determinants, will also be assessed to clarify their contribution to receptor engagement.
The results presented in Table 2; Fig. 8 indicate that the number of interactions alone is not a reliable predictor of biological activity. Notably, P6 in mice exhibits the highest number of interactions among all peptides analyzed yet fails to induce receptor activation. These findings suggest that peptide-mediated activation of the TLR4/MD-2 complex depends not merely on interaction frequency but rather on the distribution of acidic residues and interaction with specific combinations of receptor residues, distinct from those involved in LPS recognition. It is important to note that this analysis reflects the frequency of interactions observed across ten conformations of each peptide with receptor residues. While higher frequencies indicate greater potential or likelihood of interaction, they do not necessarily imply that the residue is essential for receptor activation.
To address the possibility that peptides with lower predicted binding energies could bind to serum targets, thereby reducing their effectiveness, we estimated the free energy of binding (ΔGbinding) and dissociation constant (Kd) of peptides P4, P5, and P6 using human and bovine serum albumins as representative serum protein receptors. These calculations were performed with the PRODIGY server (Table S5). P4 and P6 exhibited the strongest predicted affinity toward HSA (−14.1 and − 13.8 kcal/mol, respectively), followed by P5 (−12.7 kcal/mol). However, only P4 and P5 show biological effects, despite their high predicted albumin affinities. Consistently, the comparable nanomolar Kd values indicate similar levels of serum protein binding, suggesting that differential biological responses are unlikely to arise from peptide interaction with circulating serum proteins.
The above results indicate that strong peptide-albumin interactions alone are not sufficient to determine biological inactivity. Despite P6 showing high predicted affinity for serum proteins, it remains inactive, whereas P4 and P5 retain measurable effects even with comparable or only slightly weaker albumin binding. Therefore, the absence of activity in P6 is more plausibly related to its inability to adopt an efficient orientation or establish functionally relevant interactions at the TLR4/MD-2 interface, rather than reducing effectiveness by serum proteins.
The exploratory analysis of interaction frequencies did not show a direct correlation with peptide activity but provides a complementary perspective to identify molecular interactions. While kinetic factors such as residence time (τ) and Kd have been linked to TLR4 activity, their role is not yet definitive66,67,68,69. Future studies should integrate both interaction profiles and kinetic parameters, as current evidence remains insufficient to establish a direct relationship with biological activity, particularly for peptides. The Kd values calculated with PRODIGY further illustrate the lack of correlation between theoretical affinity and biological activity (Table S5). Although P6 exhibits the lowest predicted Kd (0.03 × 10⁻⁹ M) for the mouse TLR4/MD-2 complex, indicating the highest binding stability, it failed to elicit any biological response. In contrast, P4 and P5, with markedly higher Kd values (1.9 × 10⁻⁹ M), were the only peptides to induce receptor activation. These findings orientate us to understand how predicted affinity alone does not reliably predict the functional outcome, emphasizing the role of binding orientation and residue-specific interactions in defining biological activity.
In this regard, an exploratory free energy landscape (FEL) analysis was performed on LPS and P4 as a case study within the mouse TLR4/MD-2 complex, in order to evaluate the capacity of the aforementioned ligands to achieve structural stability during molecular dynamics (MD) simulations. The FEL was obtained by projecting the trajectories onto the RMSD and radius of gyration (Rg) coordinates, and converting the bivariate probability distribution into free energy, a well-established approach for describing biomolecular conformational ensembles70,71.
As shown in Fig. 9, panels a and b, LPS- the canonical agonist for TLR4 activation -binds deeply within the MD-2 hydrophobic pocket and induces receptor dimerization to form the active TLR4/MD2/LPS complex3 Consistent with this, the FEL of LPS reveals a dominant energy minimum centered at RMSD ≈ 0.37 nm and Rg ≈ 3.085 nm, indicating a compact and stable conformational basin that corresponds to the most energetically favorable structural state of the system.
In contrast, Fig. 9, panels c and d, shows that P4 exhibits a broader and more heterogeneous landscape, with multiple local minima distributed in the ranges of RMSD ≈ 0.20 to 0.37 nm and Rg ≈ 4.025 to 4.065 nm. This behavior is typical of ligands that lack perfect complementarity with the TLR4/MD-2 binding cavity, resulting in several low-energy conformations rather than a single deep minimum72,73. Such multiple shallow basins represent meta-stable states separated by low barriers, a hallmark of flexible binding and conformational adaptability in non-canonical ligands71.

Free-energy landscape (FEL) analysis of the mouse TLR4/MD-2 complex bound to LPS and Peptide 4 (P4). Panels a and c: Three-dimensional FEL projections constructed from RMSD and radius of gyration (Rg) reveal the conformational energy basins of the complexes with LPS and P4, respectively. Panels b and d: Corresponding two-dimensional FEL maps illustrate the location and depth of the local energy minima.
The extensive blue surface area of P4 in Fig. 9, panel d, indicates that several conformations reach structural stability during molecular dynamics simulation, consistent with the observed biological effect. In contrast, LPS exhibits smaller dark-blue regions, reflecting its high complementarity with the TLR4/MD-2 complex and the limited variability of its poses. The peptides, however, show greater conformational mobility, allowing certain orientations to establish effective interactions that can lead to receptor activation.
Overall, the convergence of interaction patterns, binding stability, and conformational landscapes indicates that specific residue interactions—rather than global binding affinity—govern the modulatory capacity of these peptides on the TLR4/MD-2 receptor. These findings highlight the relevance of interaction frequency profiles and structural dynamics in shaping receptor activation potential by molecules distinct from LPS and its analogues, opening new exploratory approaches in the discovery of TLR4/MD2 specific modulators.