
Human samples
Celiac disease was diagnosed in pediatric patients according to the ESPGHAN (European Society of Pediatric Gastroenterology Hematology and Nutrition) criteria in force at the time of recruitment, and included anti-gliadin (AGA), anti-endomysium (EMA) and anti-transglutaminase antibody (TGA) titers as well as a confirmatory small-bowel biopsy. Serum samples from peripheral blood were collected for CeD-related antibody quantification and were stored at −80 °C until use. All newly diagnosed adult CD patients had elevated TGA titers and displayed characteristic small-intestinal histopathologic abnormalities, including villous atrophy, crypt hyperplasia and intraepithelial lymphocytosis. Biopsy specimens from the distal duodenum of each individual were obtained during routine diagnosis endoscopy, after informed consent had been obtained from all subjects or their parents or legal tutors and using a protocol approved by the collaborating hospitals. None of the patients suffered from any other concomitant immunological disease. None of the controls showed small intestinal inflammation at the time of the biopsy. Information of the human samples used is summarized on Supplementary Tables 1–2.
Ethics statement
The study was approved by the Basque Country Clinical Research Ethics Board (CEIm-E ref. PI2019133) and analyses were performed after informed consent was obtained from all subjects or their parents or legal tutors. All experiments were performed in accordance with relevant guidelines and regulations.
RNA and protein extraction
RNA was extracted from human intestinal biopsies and cells with the NucleoSpin RNA Kit (Macherey Nagel, #740984.50) and proteins were purified by lysing cells in RIPA buffer (150 mM NaCl, 1.0% NP-40, 0.5% Sodium Deoxycholate, 0.1% SDS, 50 mM Tris-HCl, 1 mM EDTA).
Gene expression analyses
500–1000 ng of RNA were used for retrotranscription with the iScript cDNA Synthesis Kit (BioRad, CA, USA, #1708890). Expression values were determined by RT-qPCR using SYBR Green iTaq SYBR Green Supermix (Bio-Rad, #1725124) and specific primers. RPLP0 was used as endogenous control. Reactions were run in a BioRad CFX384 machine and melting curves were analyzed to ensure the amplification of a single product. All qPCR measurements were performed in duplicate and expression levels were analyzed using the 2–∆Ct method. All primers are listed in the Supplementary Table 4.
Anti-reovirus ELISA
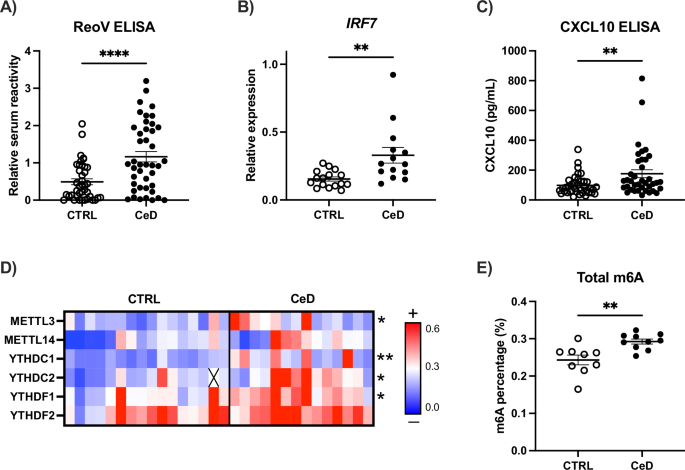
Serum reactivity against reovirus was measured by ELISA binding assay based on Amy Rosenfeld’s protocol [65], with several modifications. Briefly, ELISA was performed in flat-bottomed microtiter plates (Nunc Maxisorp ELISA plates, Thermo Fisher, #44-2404-21). Wells were coated with 105 PFU/well of Mammalian Orthoreovirus Type 1 Lang (MRV-T1L) diluted in binding buffer (0.1 M bicarbonate/carbonate buffer, pH 9.6), or with binding buffer alone as non-virus control, and incubated on a rocking platform at 4 °C overnight. The next morning, unbound virus was discarded, and wells were washed three times with PBS-Tween 20 (PBST 1X) at room temperature. Wells were blocked with 50% horse serum in PBST 1X with orbital rocking for 10 h at room temperature. After blocking, wells were washed five times before adding the serum samples, as described above. Serial 2-fold dilutions from 1:128 to 1:8192 of standard serum samples (previously pooled 3 unknown serum samples) were included in each plate to facilitate inter-plate comparisons and relativization of results. Serum samples were diluted 1:1024 in PBST 1X and wells were incubated with the samples on a rocking platform at 4 °C overnight. The following morning, sera were discarded, and wells were washed 5 times with PBST 1X on a rocking platform at room temperature for 20 min each time. Afterwards, wells were incubated with Peroxidase-conjugated AffiniPure Goat Anti-Human IgG (Jackson ImmunoResearch, #109-035-088) diluted 1:10000 in PBST 1X on a rocking platform for 90 min and washed again as described above. Serum reactivity to MRV-T1L was measured by the detection of the secondary antibody binding after addition of substrate solution BD Pharmingen TMB Substrate Reagent Set (#555214). The reaction was quenched with 2 N H2SO4 and absorbance was read at 450 nm and 570 nm.
CXCL10 and m6A ELISAs
CXCL10 concentration in serum and in cell-culture supernatants was determined by a commercially available ELISA kit (Proteintech, Germany, #KE00128) following the manufacturer’s instructions. m6A total levels were measured by commercially available m6A ELISA kit (Epigentek, NY, USA, #P-9005-96) after RNA extraction from cells cultured in different conditions.
Cell lines and treatments
Intestinal HCT-15 (#91030712) cell line was purchased from Sigma-Aldrich (Poole, UK) and cultured in RPMI (Lonza, #12-115 F) with 10% FBS, 100 units/ml penicillin and 100 μg/ml streptomycin. Cells were tested for mycoplasma every two months.
To mimic viral infections, the synthetic viral dsRNA analog polyinosine-deoxycytidylic acid (PIC) (InvivoGen, San Diego, CA, USA) was used at a final concentration of 1.5 μg/mL and transfected using Lipofectamine-2000 (Invitrogen, #11668027).
For gluten consumption simulation, cells were stimulated with pepsin-trypsin-digested gliadin (PTG) at a final concentration of 1.5 mg/mL. PTG was prepared by enzymatic digestion as described by Mendoza-Gomez et al. [66].
Combination of PIC treatment and PTG stimulation was performed as follows: (1) 300,000 cells/well were seeded and PIC was transfected when cells were still in suspension, and incubated for 24 h; (2) the next day, PIC-containing transfection medium was replaced by fresh RPMI medium; (3) the following morning PTG was added to the medium and (4) cells were harvested after 4 h of PTG stimulation.
For mRNA stability and decay evaluation, HCT-15 cells were treated with actinomycin D (Sigma-Aldrich, #A9415) at a final concentration of 5 µg/ml for 2 h, 4 h and 6 h or with cycloheximide (Sigma-Aldrich, #01810) at a final concentration of 20 µg/ml for 3 h and 24 h.
To target m6A methylation, Simvastatin (Sigma-Aldrich, #PHR1438) diluted in DMSO (Sigma-Aldrich, #D2650) was used. For in vitro experiments, HCT-15 cells were first pretreated with DMSO, and simvastatin was added at a final concentration of 10 µM after 24 h of pretreatment. Cells were finally harvested after 24 h of SV treatment. For ex vivo experiments, intestinal biopsies from CeD patients were washed in warmed wash buffer (RPMI 1640 medium + 0.1 mM DTT) and PBS 1X. Biopsies were then placed in individual wells of tissue culture plates (p96) containing 150 μL RPMI supplemented with 10% FBS, 8.0 mM L-glutamine, 3 mM Na Pyruvate, 60 mM Hepes, 100 U/mL Penicillin/Streptomycin (Pen/Strep), 0.3 unit/mL bovine insulin. Each specimen was treated with Simvastatin (Sigma-Aldrich, #PHR1438) or DMSO (Sigma-Aldrich, #D2650), and incubated up to 24 h at 37 °C (5% CO2).
Western blot analyses
Laemmli buffer 6X (62 mM Tris-HCl, 100 mM DTT, 10% glycerol, 2% SDS, 0.2 mg/mL bromophenol blue, 5% 2-mercaptoethanol) was added to protein extracts in RIPA buffer and proteins were denatured at 95 °C for 10 min. Proteins were migrated on 10% SDS-PAGE gels. Following electrophoresis, proteins were transferred onto nitrocellulose membranes using a Transblot-Turbo Transfer System (BioRad) and blocked in 5% non-fatty milk diluted in TBST 1X (20 mM Tris, 150 mM NaCl and 0.1% Tween 20) at room temperature for 1 h. The membranes were incubated overnight at 4 °C with primary antibodies at a 1:1000 dilution. Immunoreactive bands were revealed using the Clarity Max ECL Substrate (BioRad, #1705062) after incubation with a horseradish peroxidase-conjugated anti-rabbit or anti-mouse secondary antibody at a 1:10,000 dilution in 2.5% non-fat milk at room temperature for 1 h. Immunoreactive bands were detected using a Bio-Rad Molecular Imager ChemiDoc XRS (BioRad) and quantified using the ImageJ software (BioRad).
The following antibodies were used for Western Blotting: IRF7 (Santa Cruz Biotechnologies, #sc-74472), METTL3 (Abcam, #195352), ALKBH5 (Novus Biologicals, #NBP1-82188), YTHDC2 (Proteintech, #27779-1-AP) HSP90 (Cell Signaling; #4874), GAPDH (Santa Cruz Biotechnologies, #sc-47724), TUBULIN (Sigma-Aldrich, #T9026).
m6A RNA immunoprecipitation (meRIP)
Prior to the preclear and antibody-coupling steps, 10 μL and 25 μL per sample, of Dynabeads protein G (Invitrogen, # 10004D) respectively, were washed twice in reaction buffer (150 mM NaCl, 10 mM Tris-HCl, 0.1% NP-40), and resuspend in the same initial volume. For the antibody-coupling step, 1 µg of m6A antibody (Abcam, #ab151230) and control antibody (IgG, Santa Cruz Biotechnologies, Dallas, USA, #sc-2025) were coupled to 25 μL of Dynabeads protein G per sample and incubated on a rotation wheel for 2 h at 4 °C. Meanwhile, for the preclear step, first 1–5 μg of RNA per sample was fragmented with RNA fragmentation buffer (100 mM Tris, 2 mM MgCl2) for 3 min at 95 °C and placed on ice immediately after heating. Then RNA was incubated with 10 μL of Dynabeads protein G per sample and incubated in a rotation wheel for 1 h at 4 °C. After incubation, precleared RNA 10% of RNA was kept as input. Beads coupled to antibodies were washed twice in reaction buffer. RNA was added to the antibody-coupled beads and incubated for 3 h at 4 °C in a rotating wheel. Subsequently, beads were washed twice in reaction buffer, twice in low salt buffer (50 mM NaCl, 10 mM TrisHCl and 0.1% NP-40) and twice in high salt buffer (500 mM NaCl, 10 mM TrisHCl and 0.1% NP-40). After the last wash, beads were resuspended in Lysis buffer and RNA was extracted using the PureLink RNA extraction kit (Invitrogen, Carlsbad, USA, #12183016).
Gene silencing experiments
For ALKBH5, or METTL3 silencing, 2 different siRNAs against ALKBH5 (IDT, #hs.Ri.ALKBH5.13.1 and hs.Ri.ALKBH5.13.3), METTL3 (IDT, #hs.Ri.METTL3.13.1 and #hs.Ri.METTL3.13.2) or negative control siRNA (IDT, #51-01-14-01) were used.
For silencing experiments, 150,000 cells/well were seeded and 30 nM and 60 nM of siRNAs against ALKBH5 and of METTL3, respectively, were transfected into cells using Lipofectamine RNAiMax reagent (Thermo Fisher, #13778075). The following morning the siRNA-containing transfection media were replaced by fresh RPMI medium, and cells were harvested after 48 h from the change of medium.
Overexpression experiments
For METTL3 overexpression, a commercially available plasmid from Addgene (#53739) was used. For YTHDC2 overexpression, a Flag-tagged YTHDC2 plasmid cloned as previously described [67] was kindly provided by the Chuan He Group from Chicago University. For IRF7 overexpression, a CMV driven overexpression vector with coding sequence of IRF7 was purchased from VectorBuilder. For m6A-lacking IRF7 overexpression, m6A motifs were removed by mutagenesis and a MUT IRF7 plasmid was generated (described in the following section). For YTHDF2 overexpression a commercially available plasmid from Addgene (#52300) was used.
For overexpression experiments 250,000 cells/well were seeded and 500 ng plasmid was transfected using X-TremeGENE HP DNA transfection reagent (Sigma-Aldrich, #6366546001). Cells were harvested after 48 h post transfection.
IRF7 mutant plasmid construction
The MUT IRF7 plasmid (with truncated m6A motifs) was constructed in three steps. First, the region with the m6A motifs (250 bp) was amplified by PCR with specific primers containing restriction sites for BsmBI and SfiI. Afterwards, m6A motifs were removed by reamplifying the previous BsmBI/SfiI PCR product with specific primers to introduce an A→C modification of the A nucleotide of the consensus m6A motif and generate a MUT PCR product. This modification is a point silent mutation that does not affect the amino acid sequence. It neither reduces the codon usage for Glycine amino acid, based on Codon and Codon Pair Usage Tables (CoCoPUTs) (https://dnahive.fda.gov/dna.cgi?cmd=codon_usage&id=537&mode=cocoputs). Finally, this MUT PCR product was cloned into the IRF7 overexpression vector using BsmBI (NEB #R0580) and SfiI (NEB #R0123S) restriction enzymes. The primers used for mutagenesis and cloning are listed in the Supplementary Table 3.
RNA immunoprecipitation assay (RIP)
For RIP experiments, cells were lysed in RIP buffer (150 mM KCl, 25 mM Tris, 0.5 mM DTT, 0.5% NP-40, PI), kept on ice for 10 min and homogenized using a syringe. Meanwhile, 10 μL of Dynabeads protein G (Invitrogen, # 10004D) per sample were washed twice in RIP buffer and resuspend in 10 μL of RIP buffer after washes. Lysates were pre-cleared with Dynabeads protein G (Invitrogen, #10004D) for 1 h in a wheel shaker at 4 °C. After incubation, the clear supernatant was kept and separated into three aliquots (45% for target protein IP and 45% for control IP and 10% for input). The input aliquot was kept on ice or at −80 °C until further use. Pre-cleared lysates were incubated with 1 μg of the antibody of interest and anti-IgG antibody (negative control; Santa Cruz Biotechnologies, #sc-2025) for 1 h on a wheel shaker at room temperature. In the meantime, 20 μL of Dynabeads were washed as before. After 1 h incubation, Dynabeads were added to the antibody-lysate mix and samples were kept rotating for 30 min on a wheel shaker at room temperature. Afterwards, beads coupled to antibody-protein complexes were washed twice in RIP buffer, twice in low salt buffer (50 mM NaCl, 10 mM Tris-HCl, 0.1% NP-40) and twice in high salt buffer (500 mM NaCl, 10 mM Tris-HCl, 0.1% NP-40). After the washes, 70% of beads were resuspended in RNA extraction buffer and 30% were used for Western Blot.
The following antibodies were used for RIPs: METTL3 (Abcam, #195352), ALKBH5 (Novus Biologicals, #NBP1-82188), YTHDC2 (Proteintech, #27779-1-AP) HSP90 (Cell Signaling; #4874), GAPDH (Santa Cruz Biotechnologies, #sc-47724).
Organ culture experiments
Two biopsy specimens from each celiac individual at the time of diagnosis were incubated at 37 °C in RPMI media (Gibco, #21875-34) supplemented with 10% FBS (Millipore, #S0115), 4 mM L-Glutamine (Gibco, #25030-081), 1 mM sodium pyruvate (Lonza, #13-115E), 20 mM Hepes (Lonza, #17-737E), 100 U/mL penicillin/streptomycin (Lonza, #17-602E) and 0.1 U/mL bovine insulin (Sigma-Aldrich, #I0516) alone or with the addition of 10 μM simvastatin. After 24 h medium was collected and biopsies were flash frozen for further RNA and protein expression analyses.
Bioinformatic resources
SRAMP
SRAMP is an online and publicly available m6A prediction server [42]. Predictions are calculated taking into account the sequence itself and the estimated secondary structure surrounding the putative m6A site. For the prediction of m6A sites within IRF7 mRNA, its cDNA sequence was used as input, and they were analyzed using the generic option for tissue or cell selection. The secondary structure was also predicted using the same set up.
iLINCs
The signatures of GFD vs active patients (EBI_4358), active patients vs controls (GSE146190) and reovirus infected vs mock infected epithelial samples (GSE94517) were visualized using L1000FWD (https://maayanlab.cloud/l1000fwd/). Simvastatin signature was assessed among the significant drugs.
Statistical analyses
At least three biological replicates were used for in vitro experiments and over ten human samples per group, to ensure adequate power to detect relevant effects consistent with standards in the field. The data are represented as the mean and the standard error of the mean of at least three biological replicates. Mean comparisons were performed by Student’s t-test or ANOVA test assuming similar variances. All statistical tests were performed using the GraphPad Prism 9 software. Two-tailed p < 0.05 were considered statistically significant.