
You have full access to this article via your institution.
In a recent Cell paper, Wang and colleagues discovered a new type of cell death caused by contact between mitochondria and the plasma membrane, which they name mitoxyperiosis. Under Toll-like receptor (TLR) activation and metabolic stress, mTORC2 is activated, which in turn inhibits RhoA from supporting the actin network, thereby promoting mitochondrial migration to the plasma membrane; this allows for site-specific oxidative damage to the plasma membrane, causing cell lysis.
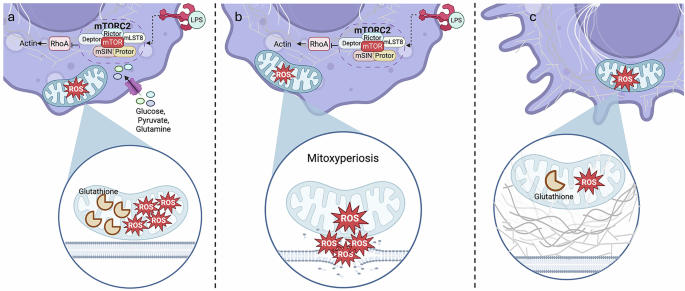
Cells can alter metabolic pathways in response to innate immune stimuli such as TLR ligands with the ensuing shift in metabolism being required for the inflammatory process.1 In the case of the TLR4 ligand LPS, this largely involves a switch to glycolysis and away from oxidative phosphorylation. Reverse electron transport occurs at Complex I generating reactive oxygen species (ROS).2 Oxidative stress has been linked to cell death, of which there are several types, including apoptosis, pyroptosis, necroptosis, ferroptosis, and PANoptosis. Wang and colleagues chose a system to study cell death which might replicate what is happening in vivo during inflammation: activation of TLRs under conditions of nutrient deprivation as often occurs at sites of inflammation.3 They termed this ‘innate immune activation and metabolic disruption’, or IIAMD. They uncovered a new type of cell death, whereby mitochondria move to the plasma membrane generating ROS, which in turn damages the plasma membrane leading to cell lysis, the type of lysis being termed mitoxyperilysis (Fig. 1). The name means that mitochondria deliver ROS to the periphery of the cell. This process, when activated in vivo, leads to tumor regression pointing to possibly new approaches to treat cancer.

a LPS stimulation drives TNF production, which promotes mTORC2 activation. RhoA is inhibited, causing a decrease in actin production, which in turn allows mitochondria to migrate to the plasma membrane. However, glutathione can mop up ROS, keeping oxidative damage under control. b LPS stimulation and glucose, glutamine and pyruvate starvation drive mTORC2 activation, which inhibits actin polymerization, driving mitochondria–plasma membrane contact. The depletion of glutathione, however, allows for uncontrollable ROS generation and membrane damage, causing mitoxyperiosis. c Under glucose, glutamine and pyruvate starvation, actin polymerization is steady, keeping the mitochondria distant from the plasma membrane. Additionally, ROS and glutathione levels are maintained at a steady state, thereby reducing oxidative damage. Created in BioRender.com.
The study began with the stimulation of murine bone marrow-derived macrophages (BMDMs) with TLR ligands under conditions of carbon starvation which comprised no glucose, pyruvate or glutamine. Only the combination induced lytic cell death, as indicated by elevated membrane rupture markers such as LDH and HMGB1. Metabolic disruption was required since supplementing glucose, glutamine or pyruvate prevented the cell death. The process was pro-inflammatory as it also promoted NLRP3 activation with subsequent production of mature IL-1β and IL-18, as well as GM-CSF, although IL-6 and TNF were unaffected. Other possible cell death mechanisms were ruled out, namely pyroptosis, apoptosis, necroptosis, PANoptosis or ferroptosis. Pore-forming mediators such as gasdermins and NINJ1 were also not involved. This suggests that cell death caused by IIAMD is unrelated to other major cell death mechanisms.
To further explore the mechanism, Wang et al. turned to metabolomics. The antioxidant glutathione was decreased after carbon starvation and LPS treatment, indicating a role for oxidative stress in IIAMD-associated cell death. This was further indicated by the inhibitory effect of N-acetyl-cysteine (NAC).
To determine the location of oxidative damage, confocal live-cell imaging was utilized to show contact between mitochondria and plasma membrane for an extended period of time in response to carbon starvation and LPS treatment. Contact resulted in membrane disruption caused by localized oxidative damage from the mitochondria, as shown by the elevated amounts of 4-hydroxy-2-nonenal, an oxidative stress marker, which was inhibited by NAC. An increase in lipid peroxidation was also observed before membrane bursting at the contact sites.
Next, BH3 domain-containing proteins known to be involved in mitochondrial damage were examined. Cell death induced by IIAMD was found to require BAX, BAK1 and BID, as was oxidative stress.
To determine the underlying mechanism of mitoxyperiosis, Wang and colleagues screened a library of 2050 small molecules, targeting ~1000 cellular pathways. They found three mTOR kinase inhibitors in the top 10 cytoprotective compounds. RNA-seq confirmed significant upregulation of the mTOR pathway during IIAMD. mTOR was then shown to be activated during IIAMD. The well-known mTOR inhibitor Torin-1 also decreased cell death, LDH release and pro-inflammatory cytokine release. By knocking down expression of mTORC1 or mTORC2, the authors found that only mTORC2 modulates and is essential for mitoxyperiosis.
Further investigation revealed that mTOR inhibition drove the formation of lamellipodia, which relocated mitochondria away from the plasma membrane. Lamellipodia are modulated by RhoA and other Rho-GTPases, impacting actin polymerization, and the authors showed inhibition of RhoA activity by IIAMD. This in turn was shown to suppress actin polymerization which allowed for prolonged mitochondria–plasma membrane contact with subsequent mitoxyperiosis.
Overall, the data show that actin networks and cytoskeletal activity inhibit mitoxyperiosis by controlling contact between the mitochondria and plasma membrane. When mTORC2 is upregulated by IIAMD, this blocks RhoA activity, inhibiting lamellipodia formation, allowing for prolonged mitochondria–membrane contact.
Wang and colleagues modeled IIAMD in mice by pre-fasting mice and then injecting them with LPS to confirm in vitro findings in vivo. These mice were shown to have higher mortality rates and tissue damage. Inhibition of mTOR reversed these IIAMD effects.
Finally, the authors examined the therapeutic potential of their findings in cancer, given how metabolism in the microenvironment can impact on anti-tumor immunity. Murine B16 melanoma cells treated in vitro with IIAMD were especially sensitive to cell death. mTOR inhibition was protective. In vivo, tumor-bearing mice were injected intratumorally with LPS in combination with fasting. Tumor size decreased while tumor necrosis increased. In addition, RhoA activity decreased in treated tumors and was restored with mTOR inhibition, confirming in vitro findings. Furthermore, necrotic tumor cells showed damaged mitochondria in proximity to the plasma membrane. These are all hallmarks of mitoxyperiosis. Activating mitoxyperiosis can therefore kill tumors, at least in the B16 melanoma model.
This work offers a new mechanism of cell death caused by the disruption of metabolism and activation of innate immunity in a process called mitoxyperiosis. When carbon-starved and TLR-stimulated, cells are depleted of glutathione, driving oxidative stress in the mitochondria. mTOR mediates the localization of mitochondria towards the plasma membrane, eventually partaking in prolonged contact. This allows for targeted oxidative damage at the contact site of the plasma membrane, degrading the lipid bilayer and causing cell lysis. Carbon starvation alone has been investigated as a cancer treatment; however, using it with LPS is a novel proposition.4 Translating mitoxyperiosis-based therapy to the clinic could serve as an effective cancer treatment. This new mechanism offers exciting revelations on how targeting immunometabolism in diseases might give rise to exciting therapeutics in cancer. Inhibiting this process could also have potential in inflammatory diseases.