
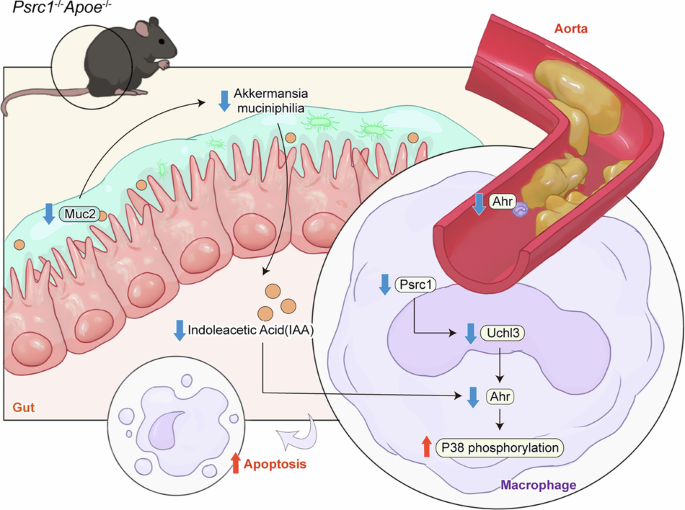
Mice and experimental design
All the animal experiments in this study were approved by the Animal Experiment Committee of Nanfang Hospital at Southern Medical University. C57BL/6J mice were purchased from Guangdong Medical Laboratory Animal Center. Apoe−/− mice were obtained from Viewsolid Biotech Co. Ltd., which were called Psrc1+/+Apoe−/−, while Psrc1−/−Apoe−/− mice were generated in the background of Apoe−/− using the CRISPR–Cas9 system via deleting exon 4, which is essential for the function of Psrc1 protein, as in our previous study8,35. Offspring from the parental lineages served as the mice model in the current study. The experimental mice were acclimatized and fed with the chow diet in the specific pathogen-free environment at the Experimental Animal Research Center of Nanfang Hospital in Southern Medical University. Eight-week-old male Psrc1+/+Apoe−/− and Psrc1−/−Apoe−/− mice were randomly assigned to distinct treatment groups prior to initiation of a 12-week atherogenic high-fat diet (HFD) feeding regimen. The subsequent experimental interventions were implemented in accordance with the specific design of each respective assay as detailed below:
Experiment 1
For the analysis of the effect and mechanism of Psrc1 knockout on Trp metabolism throughout the progression of atherogenesis, 8-week-old male Psrc1+/+Apoe−/− and Psrc1−/−Apoe−/− mice were fed a HFD (Guangdong Medical Laboratory Animal Center) for 12 weeks. Biological samples including blood, peritoneal macrophages, jejunum, ileum, colon, aorta and aortic root were systematically collected following anesthesia with isoflurane.
Experiment 2
For analysis of the effect of A. muciniphila supplementation on Psrc1 deficiency-mediated atherogenesis and reduction of IAA, 8-week-old male Psrc1+/+Apoe−/− and Psrc1−/−Apoe−/− mice were fed a HFD for 12 weeks. At week 6 of HFD initiation, mice were pretreated with a broad-spectrum antibiotic cocktail (vancomycin 0.5 g/l, neomycin 1 g/l, metronidazole 1 g/l and ampicillin 1 g/l) for 14 consecutive days. Following antibiotic clearance, Psrc1+/+Apoe−/− and Psrc1−/−Apoe−/− mice were randomly assigned to receive daily oral gavage of either 1 × 10⁹ c.f.u./ml live A. muciniphila, heat-killed A. muciniphila (treated at 121 °C for 15 min) or an equivalent volume of phosphate-buffered saline (PBS) as a control, with the intervention lasting for 28 days. One group received oral gavage with live A. muciniphila, while another group received equivalent A. muciniphila heated at 121 °C for 15 min. Biological specimens including blood, peritoneal macrophages, aorta and aortic root were systematically collected following anesthesia with isoflurane.
Experiment 3
For the analysis of the effect and mechanism of IAA supplementation on Psrc1 deficiency-mediated atherogenesis, 8-week-old male Psrc1+/+Apoe−/− and Psrc1−/−Apoe−/− mice were fed a HFD for 12 weeks. Starting from week 8 of HFD feeding, the intervention was conducted for a consecutive 28 days. Psrc1+/+Apoe−/− were randomly assigned to receive daily oral gavage of vehicle, 50 mg/kg/day IAA (MedChemExpress) or 15 mg/kg/day atorvastatin (Selleck Chemicals) as a positive control. In parallel, Psrc1−/−Apoe−/− were randomly assigned to receive equivalent vehicle, IAA monotherapy or IAA combined with 10 mg/kg/day Ahr inhibitor CH-223191 (Selleck Chemicals). For the combined treatment group, CH-223191 was administered via oral gavage 2 h before IAA supplementation. Biological specimens including blood, aorta and aortic root were systematically collected following anesthesia with isoflurane.
Experiment 4
For the analysis of the toxic effects of graded doses of IAA in Apoe−/− mice, 16-week-old male Apoe−/− mice were orally administered vehicle, 10, 50, 250 or 500 mg/kg IAA daily for 4 weeks. Blood samples and heart tissues were collected after anesthesia.
Patients
The procedures in this study were approved by the Ethics Committee of Nanfang Hospital, Southern Medical University, China (approval number NFEC-2019-185). Blood samples from 26 patients without CAD 77 patients with CAD were collected in the cardiovascular department of our hospital. The patients with CAD were diagnosed when coronary angiography revealed ≥50% stenosis in any major coronary artery or in its major branch. The patients without CAD were hospitalized for chest pain but coronary angiography indicated <50% stenosis in all major coronary artery and the branches36. Patients with heart failure, severe liver and renal dysfunction, infectious diseases, autoimmune diseases, malignancy or treated with antibiotics were excluded in this study. This study was performed according to the guidelines of Declaration of Helsinki and all the participants provided informed consent. The baseline clinical characteristics of the enrolled participants are presented in Supplementary Table 1.
Assessment of aortic atherosclerotic lesions
For analysis of the en face plaque area percentage of aorta, the aorta in mice was dissected, fixed with 4% paraformaldehyde (G1101, Servicebio) for 48 h and then stained with Oil red O (ORO; Yuanye Bio-Technology). For the analysis of the content of plaques in the aortic root, the heart of mice was dissected, fixed with 4% paraformaldehyde and embedded with OCT (Sakura) compound or paraffin. Sequential cross-sections of aortic roots were sliced, then the tissue fixed-frozen or paraffin sections were stained with hemotoxylin and eosin (HE) to detect the lesion area and necrotic cores in plaques, and the tissue fixed-frozen sections were stained with ORO to detect lipids content in plaques and Masson’s trichrome to detect the collagen content. The macrophage and smooth muscle cells in plaques were detected via immunofluorescence staining with antibodies against CD68 (97778S, CST) and α-SMA (19245S, CST), respectively. For the analysis of Psrc1 in macrophages and smooth muscle cells or Ahr in macrophages, tissue slides were stained by co-immunofluorescence with Psrc1, CD68 and α-SMA or Ahr and CD68. To detect apoptotic macrophages in plaques, tissue slides were stained by co-immunofluorescence with TUNEL (G1506, Servicebio) and CD68. The quantification was analyzed via Image J software.
Immunofluorescence staining
Tissue slides were fixed with 4% paraformaldehyde and cells in a confocal dish with methyl alcohol for 30 min, washed in PBS for 5 min and incubated with 5% BSA (w/v) in PBS for 1 h at room temperature. Tissue slides or cells were incubated with primary antibodies, such as anti-Psrc1 (GTX128047, GeneTex), anti-CD68, anti-α-SMA or anti-Ahr (28727-1-AP, Proteintech), overnight in 4 °C environment and incubated with Alex Fluor 488, Cy3 or Cy5-conjugated secondary antibodies (SeraCare Life Sciences) for 1.5 h in a dark room. Slides or cells were then incubated with 4,6-diamidino-2-phenylindole (DAPI) (D9542, Sigma-Aldrich) for staining the nucleus. Fluorescent images were captured with a TCS SP8 confocal laser scanning microscope (Leica).
Total RNA isolation and RT–qPCR analysis
The total RNA from cells or tissues was extracted with TRIzol (Thermo Fisher) reagent and was subsequently reverse transcribed to cDNA with PrimeScript RT Master Mix (Takara) according to the manufacturer’s instruction. RT–qPCR was performed with Hieff qPCR SYBR Green Master Mix (Yeasen) on a LightCycler 480 II system. Primers of target gene are listed in Table 1, and β-actin was used as control to normalize expression. Relative expression was calculated with 2−ΔΔCT method.
Determination of tryptophan metabolites in plasma
For the determination of tryptophan metabolites, 50 μl plasma was mixed with 300 μl acetonitrile containing an internal standard, followed by vortex mixing at 1000 rpm for 10 min at 4 °C. Samples were then incubated at −20 °C for 30 min to enhance metabolite extraction efficiency and subsequently centrifugated at 18,000g. The supernatant was transferred to new tubes and dried via vacuum centrifugation. Dried residues were reconstituted in 0.1% formic acid aqueous solution with vortexing, then the residual particulates were removed by additional centrifugation. Supernatant was transferred to 96-well plates and maintained at 4 °C until liquid chromatography–mass spectrometry (LC–MS) analysis. Metabolites were analyzed using an ultra-performance liquid chromatography–tandem mass spectrometry (UPLC–MS/MS) system (ACQUITY UPLC-Xevo TQS, Waters Corp.) with a scheduled system. Mobile phase A was 0.1% (v/v) formic acid in ultrapure water and mobile phase B was 0.1% (v/v) formic acid in acetonitrile. The samples were eluted by gradient program as follows: 1% B, 1 min; 10% B, 2 min; 30% B, 4 min; 70% B, 7 min; 99% B, 8.8 min; 99% B, 8.8 min; 1% B, 10 min. The flow rate was 0.40 ml/min. The injection volume was 10.0 μl. Raw UPLC––MS/MS data files were processed using MassLynx Software for metabolites peak detection, integration, calibration and quantitation. Processed datasets were imported into the iMAP (v1.0, Metabo-Profile) metabolomics platform for advanced analysis.
Cell isolation and culture
Bone marrow-derived macrophages (BMDMs) were isolated from 8–16-week-old male C57BL/6J mice. Following cervical dislocation, mice were immersed in 75% ethanol for sterilizing. After the skin removed, bilateral hindlimbs were transected at coxofemoral and talocrural articulations. The dissected specimens were maintained in PBS containing a 1% penicillin–streptomycin cocktail. The surrounding muscle tissues were removed and the femurs and tibias were transected at both ends. The bone marrow suspension was subsequently collected by flushing with Dulbecco’s modified Eagle medium (DMEM) (Thermo Fisher) containing a 1% penicillin–streptomycin cocktail (Thermo Fisher). The cellular suspension was centrifuged and ultimately reconstituted in complete culture medium containing 10% FBS (ExCell Bio), 1% penicillin–streptomycin cocktail and 20 ng/ml MCSF (GenScript) in DMEM. Cells were cultured at 37 °C in a 5% CO2-humidified incubator, with complete medium replacement conducted at 48-h intervals. Following 7-day in vitro differentiation, BMDMs were prepared for subsequent experiments.
Primary murine peritoneal macrophages (MPM) were obtained from Psrc1+/+Apoe−/−and Psrc1−/−Apoe−/− mice a fed with HFD. Peritoneal cells were collected from mice by sterile lavage and subsequently cultured in complete medium containing 10% FBS and a 1% penicillin–streptomycin cocktail in DMEM for 6–8 h. Following this incubation period, adherent cells were collected as MPM.
Human peripheral blood mononuclear cells (PBMCs) were isolated from blood samples using Ficoll-Paque Premium (17544203, Cytiva) solution-based gradient centrifugation, which differentially separates blood components according to their densities. PBMCs were washed in PBS and cultured in complete medium containing 10% FBS and 1% penicillin–streptomycin cocktail in RPMI 1640 (Thermo Fisher).
The Raw264.7 cell line was obtained from the American Type Culture Collection (ATCC). Raw264.7 was detached with culture medium, followed by centrifugation and resuspension in fresh complete medium containing 10% FBS in DMEM at 48-h intervals. MAEC was obtained from the China Center for Type Culture Collection. The MOVAS cell line was obtained from ATCC and was cultured in DMEM supplemented with 10% FBS. Cells were routinely subcultured at 3-day intervals through 0.25% trypsin–EDTA detachment for refreshing culture medium.
Determination of IAA
IAA levels in plasma samples were determined via the IAA ELISA kit (ELK7852, ELK Biotechnology) according to the manufacturer’s instructions. Standards and the samples were incubated with a biotinylated-conjugate solution for 60 min at 37 °C. The plate was washed and incubated with streptavidin–HRP for 1 h at 37 °C, followed by washing and incubating with TMB for 20 min. Stop reagent was added into plates and optical density (OD) was measured at 450 nm on an ELISA plate reader. The IAA concentrations were calculated as standard curve based on the OD of the serial dilution of IAA.
Cell transfection
The plasmid (CMV-MCS-3FLAG-SV40-EGFP) carrying the full-length coding of the mouse Psrc1 gene was subcloned into adenovirus vector as in our previous study8. BMDMs were infected with adenovirus vector control (AdGFP) or Psrc1 (AdPsrc1) for 10 h. After 10 h of infection, the cells were refreshed with complete culture medium for 48–72 h to conduct subsequent experiments. To knock down Psrc1 or cFos, BMDMs were transfected with siPsrc1 using Lipofectamine RNAiMAX (Thermo Fisher) in OptiMEM (Thermo Fisher) for 6 h. After 6 h of transfection, the medium was replaced with complete culture medium. Following 48–72 h transfection, cells were collected or used in subsequent experiments. The sequences of siCtrl, siPsrc1 and sicFos are listed in Table 2.
Bacterial strains and culture conditions
A. muciniphila (ATCC-BAA-835) was obtained from ATCC and cultured in brain–heart infusion (Thermo Fisher) supplemented with 0.5% type II mucin (Sigma-Aldrich) and 0.05% L-cysteine (Sigma-Aldrich) at 37 °C under anaerobic conditions. A. muciniphila was suspended in PBS at 1 × 109 c.f.u./ml via oral gavage administration to mice. The heat-killed A. muciniphila was heated at 121 °C for 15 min.
Total protein extraction and western blot analysis
Cell samples were lysed with RIPA buffer supplemented with a 1% protease inhibitor cocktail. The protein concentration was quantified with the BCA Protein Assay Kit (Yeasen). Equivalent protein samples were electrophoresed in SDS–PAGE gel and subsequently transferred to PVDF membranes (Millipore). Membranes were blocked in 5% skimmed milk for 1–2 h at room temperature and incubated with primary antibodies against Psrc1 (GTX128047, GeneTex), Ahr (67785-1-Ig, Proteintech), cleaved caspase 3 (9661S, CST), Bax (2772S, CST), Uchl3 (8141S, CST), phospho-P38 (4511 T, CST), total P38 (8690 T, CST), phospho-Jnk (4668 T, CST), total Jnk (66210-1-Ig, Proteintech), phospho-Erk1/2 (9101 T, CST), total Erk1/2 (9102 T, CST), Ubiquitin (3936S, CST), cFos (66590-1-Ig, Proteintech), α-Tubulin (80762-1-RR, Proteintech) and Lamin B (66095-1-Ig, Proteintech) at 4 °C overnight. Following incubation with anti-mouse or rabbit HRP-conjugated IgG secondary antibodies (SA00001-1/SA00001-2, Proteintech). The blotting bands were detected using an ECL system.
Nuclear and cytoplasmic extraction
Nuclear and cytoplasmic proteins were extracted using the NE-PER Nuclear Cytoplasmic Extraction Reagent kit (Thermo Fisher) according to the manufacturer’s protocol. BMDMs were lysed with CER I supplemented with protease inhibitor cocktail at 4 °C. After thorough vortexing to ensure uniform lysis, samples were incubated on ice for 10 min. CER II was added and samples were vortexed intermittently followed by a 1-min ice incubation. The lysates were then centrifuged at 16,000g for 5 min at 4 °C. The cytoplasmic protein in supernatant was collected. The insoluble fraction was resuspended using NER supplemented with protease inhibitor cocktail. Samples were incubated for 40 min with vigorous vortexing for 15 s every 10 min to facilitate nuclear protein release. Following incubation, samples were centrifuged at 16,000g for 10 min at 4 °C. Nuclear protein was collected in the supernatant.
Co-immunoprecipitation
Cells were washed with PBS and lysed in 10% SDS for 10 min. Protein samples were boiled for 5 min, supplemented with IP buffer (Beyotime Biotechnology) containing protease inhibitor and then centrifuged at 14,000 rpm. One out of ten samples were maintained at −80 °C as input controls, and the remaining supernatant was incubated with mouse IgG (ab18421, Abcam) or anti-Ahr antibody (sc133088, Santa Cruz Biotechnology) via mild rotation at 4 °C overnight. The magnetic beads were washed with IP buffer and incubated with protein samples containing antibody for 4 h with rotation. The magnetic beads (B23201, Selleck Chemicals) absorbing the antigen–antibody complex were magnetically isolated and washed with PBST three times. The precipitated proteins were eluted using 1× SDS–PAGE loading buffer and boiled for 10 min. The precipitated and input proteins were analyzed using western blotting.
Determination of ALT and CRE
Alanine aminotransferase (ALT) and creatinine (CRE) levels in serum samples were determined via the Alanine Aminotransferase Assay kit (Nanjing JianCheng Bioengineering Institute) and the Creatinine Assay kit (Nanjing JianCheng Bioengineering Institute) according to the manufacturer’s instructions.
Quantification and statistical analysis
All the experimental data are presented as mean ± s.d., and no outliers were excluded in this study. SPSS 29.0 was adopted for the statistical analysis. The normal distribution of the data was determined by the Shapiro–Wilk normality test. The statistical comparisons of normally distributed variables between two groups were performed using two-tailed unpaired t-tests, while the non-normally distributed variables were compared using the Mann–Whitney U test. The normally distributed variables of more than two groups were analyzed using one-way analysis of variance (ANOVA) or two-way ANOVA followed by Tukey’s or Tamhane’s T2 post hoc test, while non-normally distributed variables were analyzed using the Kruskal–Wallis H test. Rank-based inverse normal transformation of PSRC1 relative expression level, IAA level and CYP1A1 relative expression were performed, and then partial Pearson correlation coefficients were performed. The P values are shown in the figures and P < 0.05 was considered statistically significant.