
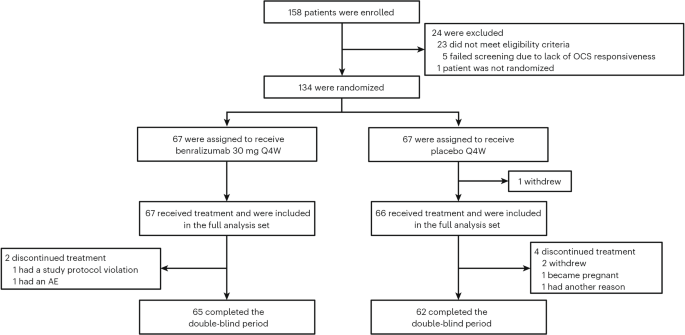
Study design
The NATRON study, a double-blind, 24-week, phase 3, randomized, placebo-controlled trial with an ongoing OLE, assessed the efficacy and safety of benralizumab in patients with HES (Supplementary Fig. 1). This study was conducted at 40 sites across 15 countries (Argentina, Austria, Belgium, China, Denmark, France, Germany, India, Israel, Japan, the Netherlands, Poland, South Korea, the UK and the USA).
All patients remained on stable background HES therapy during screening and the double-blind period; however, modifications were allowed if required for a HES flare (treatment intensification) or an AE (treatment de-escalation) thought to be due to background therapy. Stable background therapy included oral, topical, nasal or inhaled corticosteroids, immunosuppressive or cytotoxic agents, interferon-alpha (IFN-α), and other medications used to control HES and/or manage HES symptoms.
Patients who completed the 24-week double-blind period were eligible to enter an OLE where all patients received benralizumab. The end-of-study definition is described in the Supplementary Information.
The trial was conducted in accordance with the ethical principles of the Declaration of Helsinki and is consistent with the International Council for Harmonisation Good Clinical Practice guidelines, the applicable regulatory requirements and the AstraZeneca policy on bioethics. The protocol, protocol amendments and any other relevant documents were reviewed and approved by the Independent Ethics Committees/Institutional Review Boards listed in the Supplementary Information. All patients provided written informed consent. International Council for Harmonisation E6 Good Clinical Practice guidelines were followed. Patients enrolled in the study were not compensated for their participation; however, reasonable reimbursement of expenses incurred by the patients (for example, travel and parking) was provided if allowed by local regulations. This was stated clearly in the informed consent form.
An independent safety monitoring board, comprising two clinicians and a statistician, monitored overall patient safety. To inform study design and conduct, 60 patients with HES were surveyed online with support from the American Partnership for Eosinophilic Disorders patient advocacy group, Invitae and Covance Patient Engagement to provide insights on study participation, the placebo-controlled design, extended dosing, site visits and support strategies for long-term study engagement. The full protocol is publicly available at https://www.astrazenecaclinicaltrials.com/study/D3254C00001/. This trial is registered with ClinicalTrials.gov (NCT04191304) and the OLE is ongoing (closed to recruitment).
Patients were ≥12 years of age, had a diagnosis of HES (defined as history of persistent eosinophilia (>1,500 cells μl−1) without secondary cause on two examinations (≥1 month apart) and evidence of end-organ manifestations attributable to eosinophilia), were receiving documented stable HES therapy for ≥4 weeks before screening and were experiencing an HES flare at screening or had a history of ≥2 HES flares that required escalation in therapy within 12 months before screening.
Exclusion criteria were the presence of an FIP1L1::PDGFRA fusion tyrosine kinase gene rearrangement or other known imatinib-sensitive mutation; a confirmed diagnosis of EGPA or systemic mastocytosis; the presence of life-threatening HES complications, as judged by the investigator; a history of thrombotic complications, stroke or cardiac damage; a disease severity that made the patient inappropriate for inclusion; hypereosinophilia of unknown significance; a known, preexisting, clinically significant endocrine, autoimmune, metabolic, neurological, renal, gastrointestinal, hepatic, hematologic, respiratory or other systemic disorders not associated with HES that were uncontrolled with standard treatment and, in the opinion of the investigator, could have increased risk of patient safety, interfered with study outcomes or impaired completion of the study; a documented history of clinically significant cardiac damage; a known active liver disease at the time of study; a malignancy or history of malignancy within 5 years before screening; chronic or active infections requiring systemic treatment or clinically significant viral, bacterial or fungal infection within 4 weeks before visit 1; an untreated or inadequately treated helminth parasitic infection within 24 weeks before visit 1 without documented resolution; a known immunodeficiency disorder other than those attributable to OCS or HES-related therapy; a positive HIV test; any clinically significant abnormal findings on physical examination, vital signs, hematology or clinical chemistry during screening that, in the investigator’s opinion, could have posed a safety risk, influenced study results or impaired study completion; evidence of prior benralizumab treatment failure; treatment with injectable corticosteroids within 4 weeks before randomization; receipt of any investigational product within 30 days or five half-lives (whichever was longer) before visit 1 or concurrent participation in another interventional clinical study, excluding noninterventional registry or cohort studies; receipt of any marketed or investigational biologic within 4 months or five half-lives before informed consent, unless on stable background biologic therapy unlikely to interfere with safety or efficacy assessments; receipt of live attenuated vaccines within 30 days before visit 1; a history of hypersensitivity to any biologic therapy, corticosteroids or components of the investigational product; receipt of immunoglobulin or blood products within 30 days before visit 1; a known or suspected alcohol or substance abuse that could have interfered with protocol compliance; and pregnancy, breastfeeding or lactation at the time of the study. The full protocol can be accessed at https://www.astrazenecaclinicaltrials.com/study/D3254C00001/.
Following enrollment, eligible patients entered a 3-day screening period. To proceed to randomization, two criteria had to be met: first, an AEC ≥1,000 cells μl−1 at local laboratory testing on the date of enrollment, and second, a demonstration of corticosteroid responsiveness defined as an AEC <1,000 cells μl−1 after 2 days of OCS administration (1 mg kg−1 day−1 prednisone/prednisolone equivalent) given in addition to the patient’s background therapy for HES before randomization. This OCS-responsiveness assessment served as a safety measure to ensure that flares could be managed clinically without the need for investigators to monitor eosinophil counts, as eosinophil counts were blinded during the study. The OCS dose equivalency is shown in Supplementary Table 1.
Patients who met eligibility criteria at the end of the screening period were stratified by geographic region (North America, Europe, Asia and rest of the world) and HES flare status (active flare or historic flares at study entry). The inclusion of HES flare status as a stratification factor was introduced in June 2022; before this amendment, patients were required to be actively flaring at entry.
Randomization and masking
All patients were centrally assigned to a randomized study treatment using Interactive Web Response Systems (IWRS)/Interactive Voice Response Systems (IVRS). As patients became eligible for randomization, unique randomization codes were assigned sequentially in each stratum from a randomization list prepared by a computerized system provided on behalf of AstraZeneca. The randomization sequence was computer-generated centrally using a permuted block design with a fixed block size of 4 and stratified by geographic region (North America, Europe, Asia and rest of the world) and HES flare status at screening. Patients, sponsor, site staff and investigators were blinded to treatment allocation and to patients’ blood and biopsy leukocyte counts during the double-blind treatment period and up to week 4 of the OLE. All packaging and labeling ensured blinding for all sponsor and investigational site staff. The following personnel had access to the randomization list during the study: those generating the randomization list, personnel at the IWRS/IVRS company, AstraZeneca’s supply chain department, drug safety services representatives (data entry site case handlers), the bioanalytical laboratory performing the PK sample analysis, the independent statistical data analysis center, the unblinded programmer and the unblinded medical monitor. The IWRS/IVRS provided the investigators with the kit identification number to be allocated to the patient at the dispensing visit.
Procedures
Patients were randomly assigned 1:1 into the 24-week double-blind period where they received benralizumab 30 mg (accessorized prefilled syringe) subcutaneously every 4 weeks or a matching placebo, in addition to the patient’s background therapy for HES. The rationale for benralizumab dosing is described in the Supplementary Information.
In addition to screening and scheduled study visits every 4 weeks, patients were advised to contact the study site each time they thought their symptoms were worsening and attend the site for a flare visit assessment. If study treatment was discontinued early, patients attended a discontinuation visit 4 ± 1 weeks after the last dose.
HES flares were assessed by the investigator at all scheduled or flare visits. Vital signs and blood samples were collected at screening and all scheduled and flare site visits; for scheduled dosing visits, samples were taken before the administration of study treatment.
Patient-reported outcome assessments were completed by patients using an electronic device at study site visits before other study procedures. Patient-reported outcome assessments included: (1) PROMIS Fatigue short form 7a, measured at screening and scheduled study visits (that is, every 4 weeks); (2) SF-36v2 (acute recall), measured at screening, visit 6 (week 12) and visit 9 (week 24); (3) PGI-S, measured at screening and scheduled study visits (every 4 weeks); and (4) PGI-C, measured at visit 4 (week 4) and scheduled study visits (every 4 weeks).
PK and immunogenicity assessments were conducted at screening and pre-dose at scheduled visits 4 (week 4), 5 (week 8), 7 (week 16) and 9 (week 24). AEs included events reported between screening (visit 1) and last contact with the patient. Serious AEs included events recorded from written informed consent throughout the duration of the study.
Outcomes
The primary endpoint was time to first HES flare during the 24-week, double-blind treatment period. A flare was defined as HES clinical manifestation or laboratory abnormality resulting in an increase of OCS ≥10 mg day−1 prednisone equivalent for ≥2 days, or an increase or addition of a new cytotoxic and/or immunosuppressive therapy, or hospitalization. Flares were assessed by the investigator through complete or brief physical examinations, an investigator-led HES symptom interview, laboratory assessments and other routine safety assessments; please refer to the Supplementary Information for further details. If patients were unable to attend the study site for flare assessment, medical records were collected and an investigator-led HES symptoms interview was recommended. Time to first HES flare was calculated as the number of days from the date of randomization to the start date of the first flare event, plus 1 day. The start date of HES flare was defined as the first day of increased dose/burst of OCS, first day of any increase or addition of new cytotoxic and/or immunosuppressive therapy, or date of hospital admission, whichever occurred first.
Secondary endpoints that were multiplicity protected within the prespecified statistical testing hierarchy were defined as ‘key’. Key secondary endpoints were: (1) the proportion of patients with HES flares, with those who withdrew from the study without having experienced a flare considered as having had a flare event; (2) the annualized rate of HES flares, assessed over a maximum follow-up period of 24 weeks or, for patients lost to follow-up, the follow-up time was defined as the duration from randomization to the last timepoint at which flare status could be evaluated, with distinct flares defined as those with onset occurring ≥14 days after the resolution of the previous flare; (3) the time to first hematologic relapse (AEC ≥1,000 cells μl−1), calculated as the number of days from the date of randomization to the start date of first hematologic relapse plus 1 day; and (4) the change from baseline to week 24 in PROMIS Fatigue, with a standardized total score calculated for each visit over the double-blind period.
Other secondary endpoints included the proportion of patients with hematologic relapse (including those who withdrew from the study) during the double-blind period, the proportion of patients with AEC <500 cells µl−1 for 24 weeks, the proportion of patients requiring an increase in corticosteroid dose at any time during the double-blind period and other patient-reported outcomes (SF-36v2, PGI-S and PGI-C).
PK was assessed through benralizumab serum concentrations and immunogenicity assessed through ADA assays and neutralizing antibody testing, as previously described29. Safety was assessed through reporting of AEs, serious AEs, vital signs and clinical laboratory variables.
Statistical analysis
All efficacy endpoints, demographics and baseline characteristics were analyzed using the full analysis set, which included all randomized patients who received ≥1 dose of study treatment according to the intention-to-treat principle, irrespective of adherence to the protocol and continued trial participation.
The primary analysis data cutoff was to occur after 38 patients had a first HES flare event and all randomized patients had completed the 24-week double-blind period. It was estimated that approximately 38 first HES flare events during the double-blind period were required to detect a statistically significant difference between treatment groups at the two-sided 5% significance level with approximately 80% power if the true treatment effect is an HR of 0.389 (equivalent to 30% of patients receiving benralizumab experiencing an event by the end of the double-blind period versus 60% of patients receiving placebo). On the basis of these assumptions, a sample size of approximately 120 was expected, although recruitment could continue beyond this to provide confidence that sufficient events would be observed once complete follow-up was achieved. A sensitivity analysis was conducted to assess the impact of patients changing systemic background therapy before HES flares, by censoring any patients with systemic OCS or immunosuppressive therapy changes that were considered to have a potential to impact the chance of the patient flaring. Please refer to the Supplementary Information for further details on the sensitivity analysis. Subgroups were analyzed for the primary endpoint, and included sex, age, region (a stratification factor), race, HES subtype (based on investigator assignment), baseline blood eosinophil counts, baseline OCS dose, HES flare status (a stratification factor), primary organ involvement and time since HES diagnosis.
To account for multiplicity, if the primary endpoint was found to be statistically significant, key secondary endpoints were tested using a hierarchical fixed sequence approach at two-sided 0.05 in the following order: (1) the proportion of patients who experience a HES flare during the double-blind period; (2) the number of HES flares (annualized rate) during the double-blind period; (3) the time to first hematologic relapse during the double-blind period; and (4) the change from baseline in PROMIS Fatigue score at week 24.
Time-to-event endpoints were analyzed using a stratified log-rank test, adjusted for region. HRs and 95% CIs were estimated using a Cox proportional hazards model with treatment group and region as covariates. For patients who had not experienced the event, the time to event was censored at the end of the double-blind period corresponding to the date of the first of benralizumab open-label dose, study day 183, the date of last contact or the data cutoff date, whichever occurred first.
The proportion of patients with a HES flare was analyzed using a logistic regression model with treatment group and region as covariates to obtain the OR and 95% CIs (withdrawals were included as flare events in the primary analysis). The annualized rate of flares during the double-blind period was analyzed using a negative binomial model. The logarithm of the follow-up time was used as an offset variable in the model. The model included covariates of treatment group and region to obtain estimates of the flare rates in each treatment group and the rate ratio for benralizumab versus placebo.
For continuous change from baseline endpoints, including changes in PROMIS Fatigue standardized T-scores, data were analyzed using a mixed model for repeated measures, with treatment group, baseline score, visit, region and treatment visit interaction as covariates. No imputations were performed for missing data; all available observations were included in the analyses, which assumed missing data were missing at random. Safety was analyzed descriptively on the basis of the safety analysis set, which included all patients who received ≥1 dose of study treatment. The PK analysis set included all patients who received ≥1 dose of benralizumab with ≥1 quantifiable serum PK observation post-first dose. All data analyses were performed with SAS System (SAS Institute Inc.) software. Please refer to the Supplementary Information for additional details on statistical analyses.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.