
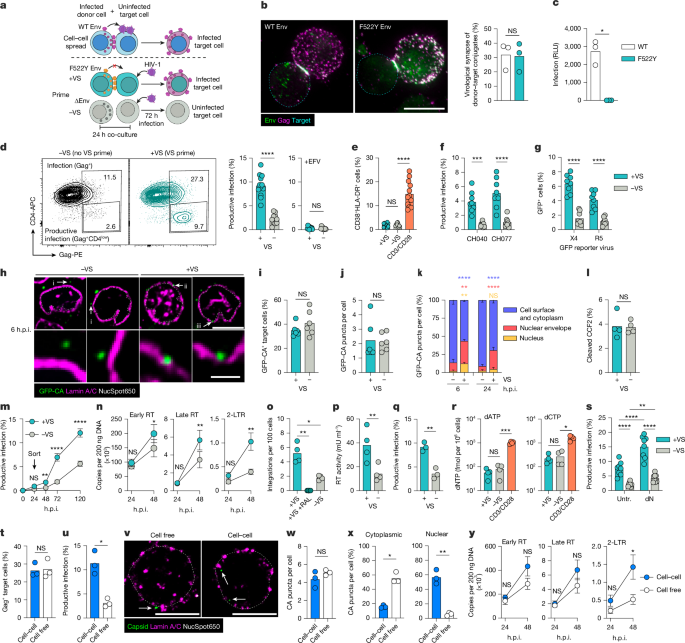
Cells
Peripheral blood mononuclear cells (PBMCs) were isolated from leukocyte cones from healthy donors (NHS Blood and Transplant Services) by density centrifugation using Lympho 24+ Spin medium (PluriSelect) and pluriMate II tubes (PluriSelect). PBMCs were cryopreserved in 10% DMSO (Sigma-Aldrich) in FCS (LabTech). CD4+ T cells were isolated by negative selection using the MojoSort Human CD4 T cell Isolation Kit (BioLegend) and cultured in RPMI1460 medium supplemented with 10% FCS, 1% penicillin–streptomycin and 1% GlutaMax (Thermo Fisher Scientific) (complete RPMI medium) with 10 IU ml−1 IL-2 (Center for AIDS Reagents (CFAR), National Institutes of Biological Standard and Control, UK). For CD25/CD69 depletions biotin-Ab cocktail from the MojoSort Human CD4 T cell isolation kit was supplemented with biotin–anti-CD25 (M-A251, BioLegend) and biotin–anti-CD69 (FN50, BioLegend) antibodies (both at 10 μg per 50 million cells) to retain CD25/CD69 expressing cells in the negative fraction. Purity was assessed by flow cytometry (described below). Isolated CD4 T cells (8–12 × 106 cells) were activated in Nunclon Delta T25 flasks (Thermo Fisher Scientific) using plate-bound anti-CD3 antibody (OKT3, 5 μg per flask, BioLegend) and soluble anti-CD28 antibody (CD28.2, 2 μg ml−1, BioLegend). After 3 days of activation, cells were cultured/rested in fresh medium (without CD3/CD28 antibodies) for another 2 days before being used for infections or co-cultures. Resting purified CD4 T cells were cultured in presence of 10 IU ml−1 IL-2 without CD3/CD28 activation. Jurkat T cell lines clone E6-1 (ATCC TIB-152) and clone 1G5 (containing Tat-driven luciferase reporter, obtained from AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH) were cultured in complete RPMI medium. HEK293T/17 cells (ATCC, CRL-11268) and HeLa-TZMbl cells (CFAR) were cultured in DMEM GlutaMax medium (Thermo Fisher Scientific) supplemented with 10% FCS and 1% penicillin–streptomycin.
Plasmids
pNL4.3 plasmid (donated by M. Martin) was from the CFAR. pNL4.3 ΔEnv and pNL4.3 ΔVpr was provided by R. Sloan52. pCH040 and pCH077 were provided by G. Towers and were originally obtained through the NIH AIDS Reagent Program from J. Kappes and C. Ochsenbauer. pNLENG1-IRES NL4.3 Env and pNLENG1-IRES YU2 Env were provided by D. Levy53. pHGFP-GFPCA was generated previously and provided by V. Pathak and encodes GFP fused to CA in the NL4.3 ΔEnv backbone20. Parental pHGFP plasmid contains WT CA sequence20. pHGFP-mStayGold-CA was generated by replacing GFP sequence from pHGFP-GFPCA plasmid between the MluI and XbaI restriction sites with the mStayGold sequence cloned from the pRSETB/mStayGold plasmid (Addgene, 212017) using the following primers 5′-TCAGTGACGCGTATGGTGTCTACAGGCGAGGAGC and 5′-TCAGTCTCTAGACAGGTGGGCCTCCAGGGTC. pHGFP-Spot-tag-CA was generated by replacing the GFP sequence from the pHGFP-GFPCA plasmid between the MluI and XbaI restriction sites with the following primers also digested with MluI and XbaI enzymes: 5′-CGCGTCCAGACCGCGTGCGCGCCGTGAGCCATTGGAGCAGCGGCGGAT and 5′-CTAGATCCGCCGCTGCTCCAATGGCTCACGGCGCGCACGCGGTCTGGA. pBLAM-Vpr was obtained from Addgene and pAdVantage from Promega. pMDG expressing VSV glycoprotein was from Genscript. pWEAU_d15_410_5017 expressing HIV-1 WEAU isolate Env and pFurin were provided by L. McCoy. pCAT001 expressing codon-optimized HIV-1 HXB2 Env was obtained from Addgene (171061) NL4.3 Env-F522Y described previously17 was generated by site-directed mutagenesis using the QuickChange Lightning kit (Agilent) and the following primers: F522Y forward: 5′-GCTTTGTTCCTTGGGTAT TTGGGAGCAGCAGG; and F522Y reverse: 5′-CCTGCTGCTCCCAAA TACCCAAGGAACAAAGC.
Viruses
Viruses were made by transfecting 293T cells (7 × 106 cells seeded per T175 flask) with 10 μg HIV-1 plasmid using Fugene6 (Promega) in 1:3 ratio. HIV-1 NL4.3 ΔEnv and Env-F522Y virions were pseudotyped with Env from the primary isolate transmitter/founder virus WEAU by co-transfecting with pWEAU Env (5 μg) and pFurin (2.5 μg) plasmids. VSVg-pseudotyped NL4.3 ΔEnv and Env-F522Y were co-transfected with pMDG (2.5 μg). Blam-Vpr virus was made by co-transfecting pNL4.3 ΔVpr (10 μg), pBLAM-Vpr (2.5 μg) and pAdVantage (2 μg). GFP–CA virus was made by co-transfecting pNL4.3 WT (10 μg) with pHGFP-GFPCA (1 μg) as described previously20. mStayGold-CA virus was made by co-transfecting pHGFP (NL4.3 ΔEnv, 12 μg) with pHGFP-mStayGold-CA (1 μg) and pCAT001 HXB2 Env (1 μg) and pFurin (2.5 μg). Spot-CA virus was made by co-transfecting pHGFP (NL4.3 ΔEnv, 12 μg) with pHGFP-Spot-tag-CA (1 μg) and pCAT001 HXB2 Env (1 μg) and pFurin (2.5 μg). Viral supernatants were collected 48 h and 72 h after transfection, filtered through 0.45-μm syringe-driven filter, purified and concentrated by ultracentrifugation through 20% sucrose cushion and resuspended in complete RPMI medium. For experiments involving quantitative PCR (qPCR) analysis of viral DNA, the product supernatants were treated with DNase I (Sigma-Aldrich) for 1 h at 37 °C before ultracentrifugation. Viral titres were measured by quantifying supernatant RT activity by SG-PERT assay54. Alternatively, and where indicated, quantification of infectivity (TCID50 per ml) was determined by titration of viral supernatants on HeLa-TZMbl reporter cells using Luciferase Bright-Glo substrate (Promega) and GloMax luminometer (Promega).
HIV-1 infections, VS priming and bead priming
Donor cell infection
Purified primary activated CD4 T cells (donors) were infected on day 5 after activation by spinoculation (2 h, 1,200g, 25 °C) with Env-pseudotyped NL4.3 Env-F522Y virus or NL4.3 ΔEnv virus (1.0 and 0.5 U RT per 106 cells, respectively) in the presence of DEAE-dextran (0.1 μg per 106 cells). Jurkat donor cells were infected with VSV-g pseudotyped NL4.3 Env-F522Y and NL4.3 ΔEnv virus (0.5U RT per 106 cells) by gravity infection for 4 h before changing the medium. Infected donor cells were incubated for 2 days before use, and infection was confirmed by flow cytometry.
VS priming
Target T cells (resting or activated) were prelabelled with 2.5 μM eFluor 450 cell proliferation dye (Thermo Fisher Scientific) according to the manufacturer’s instructions. Dye-labelled resting target T cells were then co-cultured with autologous primary donor T cells at a 1:1 ratio for 24 h and challenged with NL4.3 WT, NL4.3 GFP–CA, CH040, CH077 (1 U RT per 106 cells unless otherwise indicated) or CXCR4- and CCR5-tropic GFP reporter viruses (2 U RT per 106 cells). Infection was measured 72 h later (unless otherwise indicated) by flow cytometry staining for cell surface CD4 and intracellular viral Gag (described below). Alternatively, for activated target T cell experiments, primary target T cells were used at day 5 after activation and co-cultured with either HIV-1 infected autologous activated donor cells or Jurkat donor cells. After 24 h, VS-primed targets were challenged with second virus as above, and infection was measured 24 h later.
Bead priming
Polystyrene streptavidin-coated beads with 5 μm diameter (Bangs Laboratories) were coated with biotin-tagged anti-CD4 antibody (OKT4, BioLegend), biotin-tagged anti-CD3 antibody (OKT3, BioLegend) or biotin tagged BG505 SOSIP Env (gift from L. McCoy) according to the manufacturer’s instructions. For 25% bead binding capacity (equivalent to 7 × 105 biotin molecules per bead), 5 × 106 beads were coated with 1 μg anti-CD4 or anti-CD3 antibody or 2.5 μg SOSIP Env. Unless otherwise indicated, beads were coated at 25% binding capacity. Streptavidin-coated nanobeads with 130 nm diameter (BioLegend) were coated in excess anti-CD4 antibody or SOSIP Env (25 μg ml−1) to achieve 100% binding capacity. Target T cells (resting or activated) were incubated with beads for 1 h at 1:1 ratio (unless otherwise indicated) before infection as described above.
CCS assay
Activated primary donor cells were infected with WT NL4.3 virus (0.5 U RT per 106 cells) for 2 days as described above. Resting target cells were labelled with eFluor 450 Cell Proliferation Dye as described above and mixed with infected donor cells in 1:1 ratio (total 2 × 105 cells per well). For cell-free infection comparison, 2 × 105 target cells per well were infected with WT NL4.3 (2 U RT per 106 cells), which resulted in equal target cell infection levels at 6 h.p.i., compared to CCS 6 h after coculture as measured by flow cytometry and immunofluorescence (described below).
For qPCR analysis, approximately 5 × 105 target cells were sorted from co-cultures as described below. For inhibitor experiments, resting target T cells were either untreated or pretreated with LCK and CDK1/2 signalling pathway inhibitors and mixed with donor cells to allow for virus CCS. After 8 h and 24 h of coculture, neutralizing anti-Env antibody (VRC01, 20 μg ml−1, CFAR) and fusion inhibitor (Enfuvirtide [T20] 0.1 μM, CFAR) were added to block further HIV-1 CCS and infection of target T cells was measured after 72 h of co-culture by flow cytometry.
Jurkat 1G5 CCS assay
Jurkat T cells (WT, E6-1 clone) were HIV-1 infected as described above and co-cultured in a 1:1 ratio with uninfected 1G5 Jurkat target T cells. Luciferase expression was measured after 24 h of coculture using Luciferase Bright-Glo substrate and GloMax luminometer.
Blam-Vpr assay
Cells were infected with NL4.3 Blam-Vpr virus (1 U RT per 106 cells) for 4 h and β-lactamase activity was measured using the LiveBLAzer CCF2-AM Kit (Thermo Fisher Scientific) as described previously55.
Inhibitors
Inhibitors were added to cells as indicated, either 1 h before co-cultures or 1 h before adding virus and used at the following concentrations. EFV (HIV RT inhibitor, 5 μM), maraviroc (CCR5 antagonist, 0.2 μM), AMD3100 (CXCR4 antagonist, 0.5 μM) and raltegravir (HIV integrase inhibitor, 5 μM) were purchased from CFAR. AB1010 (LCK inhibitor, 2 μM), IPA-3 (PAK1 inhibitor, 5 μM) and THZ1 (CDK7 inhibitor, 0.05 μM) were purchased from ApexBio. U0126 (ERK inhibitor, 4 μM), SP 600125 (JNK inhibitor, 5 μM), AKT INH VIII (AKT inhibitor, 0.1 μM), sotrastaurin (PKC inhibitor, 4 μM), MLN8237 (Aurora A inhibitor, 0.25 μM), GSK461364 (PLK1 inhibitor, 0.8 μM), NSC 663284 (CDC25 inhibitor, 4 μM), BMS-265246 (CDK1/2 inhibitor, 2 μM), palbociclib (CDK4/6 inhibitor, 1 μM) and eltanexor (exportin-1 inhibitor, 0.5 μM) were purchased from Cayman Chemicals.
T cell signalling
To measure T cell signalling during VS-priming, donor and target T cells were stained with Zombie Aqua Live/Dead dye (BioLegend) and chilled on ice before being mixed and incubated on ice for 20 min to allow for donor–target conjugate formation. Similarly, bead and target T cells were incubated on ice to allow for bead–target conjugate formation. Co-cultures were then incubated in a 37 °C water bath for 0–25 min, before being fixed with warm fixation buffer (4% formaldehyde in PBS) to arrest signalling. Signalling activation was measured by flow cytometry using phospho-specific antibodies (described below).
Flow cytometry
Cells were washed in PBS and stained with Zombie Aqua or UV Live/Dead dye (1:500, BioLegend) and antibodies against surface markers (detailed below) in PBS for 15 min at room temperature. Cells were then washed in PBS and fixed in 4% formaldehyde in PBS for 30 min. When only intracellular Gag expression was analysed, cells were permeabilized in intracellular staining permeabilization wash buffer (BioLegend) for 10 min and stained with anti-Gag antibody (KC57, PE (1:350) or FITC (1:150), Beckman Coulter) for 20 min and washed. When co-staining with other intracellular markers, the cells were permeabilized with 0.25% Triton X-100 (Sigma-Aldrich) in PBS for 15 min, blocked with 3% BSA (Sigma-Aldrich) and 0.1% Triton X-100 in PBS for 45 min, washed in PBS and stained in antibody diluted in 1% BSA in PBS for 30 min. Unconjugated primary antibodies were detected using the following secondary antibodies: anti-rabbit IgG (Poly4064, BV421 or PE, 1:200) or anti-mouse IgG (Poly4053, PE or Alexa647, 1:200) from BioLegend. For phospho-CDK1 Tyr15 staining, cells were first stained phospho-CDK1 Tyr15 followed by anti-mouse secondary antibody. Cells were then washed and incubated with additional phospho-CDK1 Tyr15 to block any excess secondary antibody binding sites and then surface stained with fluorophore-conjugated mouse anti-CD3, anti-CD4 and anti-CD8.
The following surface antigen antibodies were used: CD3 (UCHT1, FITC (1:100) or BV711 (1:250), BioLegend), CD4 (RPA-T4, APC (1:200) or APC-Fire750 (1:100), BioLegend) CD8 (SK1, BV605, BioLegend, 1:250), CD45RO (UCHL1, PerCP-Cy5.5, BioLegend, 1:150), CD69 (FN50, APC-Fire750, BioLegend, 1:100), CD25 (M-A251, PE-Dazzle594, BioLegend, 1:200), CD38 (HIT2, BV510, BioLegend, 1:150), HLA-DR (L243, BV785, BioLegend, 1:150), PD-1 (EH12.2H7, PE, BioLegend, 1:200), CD98 (REA387, PE-Vio770, Miltenyi Biotec, 1:150).
The following intracellular antigen antibodies were used: phospho-LCK Tyr394 (A18002D, PE, BioLegend, 1:150), phospho-LCK Tyr505 (REA673, APC, Miltenyi Biotec, 1:150), phospho-Zap70 Tyr319 (1503310, Pe-Cy7, BioLegend, 1:150), phospho-Zap70 Tyr292 (REA41, PE, Miltenyi Biotec, 1:150), phospho-ERK Thr202/Tyr204 (6B8B69, Alexa647, BioLegend, 1:150), phospho-AKT Ser473 (M89-61, PE-CF594, BD, 1:150), phospho-CDK1 Thr14 (A20004B, Alexa647, BioLegend, 1:150), phospho-CDK1 Tyr15 (A21009D, BioLegend, 1:200), phospho-CDK1 Thr161 (9114, Cell Signaling Technology, 1:200), total CDK1 (9112, Cell Signaling Technology, 1:200; or 2A11E4, CoraLite Plus 647, Proteintech, 1:250) phospho-Wee1 Ser642 (D47G5, Cell Signaling Technology, 1:200), total Wee (D10D2, Cell Signaling Technology, 1:200), Ki-67 (PE, BioLegend, 1:150), cyclin B1 (V152, Alexa647, BioLegend, 1:150), Glut1 (EPR3915, Abcam, 1:600), RRM1 (D12F12, Cell Signaling Technology, 1:250), RRM2 (E7Y9J, Cell Signaling Technology, 1:250), RRM2B (EPR8816, Abcam, 1:300) and HA tag (16B12, APC, BioLegend, 1:150). For EdU incorporation, cells were pulse labelled with 5 μM EdU for 3 h before Live/Dead stain and fixation. Cells were permeabilized and incorporated EdU was click-labelled with Cy3 dye using the EdU Cy3 Imaging Kit (ApexBio) according to the manufacturer’s instructions. After click-labelling, cells were washed in PBS and stained for surface antigens (CD3, CD4, CD8) as described above. Cell proliferation was measured by staining cells with 5 μM eFluor450 cell proliferation dye and assessing the decrease in eFluor450 fluorescence after 72 h compared with unstimulated cells. All of the samples were analysed on the BD Fortessa or BD Fortessa X20 (BD) system using BD FACSDiva Software v.9.0 (BD) and analysed by FlowJo v.10 (BD).
Live-cell sorting
To isolate donor and target T cells after co-culture by flow sorting, donor cells were prelabelled with CellTrace Far Red dye (Thermo Fisher Scientific) and target T cells prelabelled with eFluor450 Cell Proliferation Dye. Resting target T cells were co-cultured with activated donor cells for 24 h to allow for VS-priming and then infected for 24 h as described above. Cells were then stained with Zombie Green Live/Dead dye and passed through 20 μm cell strainer before sorting for the live target T cell population on MACSQuant Tyto (Miltenyi Biotec) cell sorter using high-speed cartridges. The purity of sorted target T cell populations was confirmed to be >99% pure by flow cytometry analysis. Sorted resting target T cells were then either returned to culture or collected immediately for analysis. Alternatively, resting or activated target T cells were co-cultured with activated donor cells for 1 h or 24 h as indicated and sorted before infection.
CDK1 mRNA electroporation
Active (T14F/Y15F) and inactive (T14F/Y15F/T161A) CDK1 mutant sequences were HA-tagged at the N-terminus, codon optimized and synthesized as gBlocks (IDT) with following sequences: active: 5′-TATCCTTACGACGTGCCCGACTACGCAGGGGGCAGTGGCGGTATGGAGGAC TATACGAAGATCGAGAAAATTGGAGAGGGCGCTTTCGGCGTTGTATACAAAGGGAGACACAAGACTACAGGACAGGTG GTAGCAATGAAGAAAATTCGCCTTGAATCTGAGGAAGAGGGGGTACCCTCCACGGCGATTCGGGAAATCTCACTCCTCAAAGAACTGCGACACCCAAATATCGTATCACTTCAGGACGTTCTCATGCAAGATAGTAGACTGTACCTCATATTCGAGTTCTTGTCAATGGATTTGAAAAAATATCTTGATAGTATCCCACCAGGCCAATACATGGACAGTTCCCTGGTGAAGTCTTATCTCTACCAGATTCTGCAAGGTATCGTCTTTTGCCACTCCCGGAGGGTATTGCACAGAGACCTCAAGCCCCAAAACCTTCTGATTGACGACAAGGGGACCATAAAGTTGGCGGACTTTGGCCTGGCTAGGGCATTTGGTATACCAATTCGGGTTTATACCCACGAAGTAGTAACACTGTGGTACAGGAGCCCCGAGGTCCTGCTCGGCTCAGCGAGGTATTCAACCCCGGTGGACATCTGGAGCATTGGCACTATTTTTGCAGAACTCGCAACTAAGAAGCCTCTTTTTCACGGCGATTCCGAAATTGATCAACTCTTCCGAATTTTTCGGGCATTGGGCACGCCTAACAACGAGGTCTGGCCGGAGGTGGAGAGTTTGCAGGATTACAAGAATACGTTTCCGAAATGGAAACCTGGCAGCCTTGCCAGCCATGTGAAGAACCTCGATGAAAATGGCCTTGACCTCTTGTCCAAGATGCTTATCTACGACCCAGCGAAACGAATCAGCGGGAAGATGGCCCTGAACCATCCCTACTTCAATGACCTGGACAACCAGATCAAGAAGATGTGA; inactive 5′-TATCCTTACGACGTGCCCGACTACGCAGGGGGCAGTGGCGGTATGGAGGACTATACGAAGATCGAGAAAATTGGAGAGGGCGCTTTCGGCGTTGTATACAAAGGGAGACACAAGACTACAGG ACAGGTGGTAGCAATGAAGAAAATTCGCCTTGAATCTGAGGAAGAGGGGGTACCCTCCACGGCGATTCGGGAAATCTCACTCCTCAAAGAACTGCGACACCCAAATATCGTATCACTTCAGGACGTTCTCATGCAAGATAGTAGACTGTACCTCATATTCGAGTTCTTGTCAATGGATTTGAAAAAATATCTTGATAGTATCCCACCAGGCCAATACATGGACAGTTCCCTGGTGAAGTCTTATCTCTACCAGATTCTGCAAGGTATCGTCTTTTGCCACTCCCGGAGGGTATTGCACAGAGACCTCAAGCCCCAAAACCTTCTGATTGACGACAAGGGGACCATAAAGTTGGCGGACTTTGGCCTGGCTAGGGCATTTGGTATACCAATTCGGGTTTATGCACACGAAGTAGTAACACTGTGGTACAGGAGCCCCGAGGTCCTGCTCGGCTCAGCGAGGTATTCAACCCCGGTGGACATCTGGAGCATTGGCACTATTTTTGCAGAACTCGCAACTAAGAAGCCTCTTTTTCACGGCGATTCCGAAATTGATCAACTCTTCCGAATTTTTCGGGCATTGGGCACGCCTAACAACGAGGTCTGGCCGGAGGTGGAGAGTTTGCAGGATTACAAGAATACGTTTCCGAAATGGAAACCTGGCAGCCTTGCCAGCCATGTGAAGAACCTCGATGAAAATGGCCTTGACCTCTTGTCCAAGATGCTTATCTACGACCCAGCGAAACGAATCAGCGGGAAGATGGCCCTGAACCATCCCTACTTCAATGACCTGGACAACCAGATCAAGAAGATGTG.
The pCDNA3.1 vector was linearized by PCR using following primers: 5′-TGACCTGGACAACCAGATCAAGAAGATGTGAGGGCCCGTTTAAACCCGCTGATC and 5′-CCCTGCGTAGTCGGGCACGTCGTAAGGATACATGGTGGCCATGCTAGCCAGC. CDK1 sequences were then inserted using the HiFi DNA Assembly kit (NEB). For in vitro transcription, CDK1 vectors were first linearized by PCR using following primers: 5′-CATGGTGATGCGGTTTTGGCAGTACATCAATGG and 5′-CAGAAGCCATAGAGCCCACCGCATCC. RNA was then transcribed, capped and polyadenylated using the HiScribe T7 ARCA mRNA Kit with tailing (NEB) according to manufacturer’s instructions and purified using the Monarch RNA cleanup kit (NEB). Resting CD4 T cells were electroporated with mRNA using the Neon Transfection System (Thermo Fisher Scientific). In brief, 1.5 × 106 cells were resuspended in 100 μl buffer T with 0–6 μg mRNA per 106 cells and electroporated with 1 pulse at 2,200 V for 20 ms. Cells were infected or collected for immunofluorescence analysis (described below) at 8 h after electroporation and CDK1 expression was monitored by flow cytometry using HA antibodies (described above).
CDK1 CRISPR–Cas9 knockout
The method for CRISPR–Cas9 knockouts in resting CD4 T cells was described previously56. In brief, lyophilized tracrRNA and crRNA (IDT) were resuspended at 160 μM in IDT Duplex buffer. Non-targeting control (NTC) and CDK1-targeting cRNAs contained GGGTTCCTAGTACTGCAATT and CCATACCCATTGACTAACTA sequences, respectively. Single-stranded donor oligonucleotides (ssODN, sequence: TTAGCTCTGTTTACGTCCCAGCGGGCATGAGAGTAACAAGAGGGTGTGGTAATATTACGGTACCGAGCACTATCGATACAATATGTGTCATACGGACACG, IDT) were also resuspended at 160 μM in IDT Duplex buffer. To generate Cas9–RNPs 1.5 μl of each tracrRNA and crRNA was mixed and incubated at 37 °C for 30 min. Then, 1 μl of ssODN was added and incubated at 37 °C for 5 min. Next, 5 μl of 20 μM EnGen Spy Cas9 HF1 (NEB) was added and incubated for further 15 min at 37 °C. Meanwhile, 1.5 × 106 CD4 T cells were washed and resuspended in 95 μl buffer T, mixed with 9 μl Cas9-RNP solution and electroporated (1 pulse, 2,200 V, 20 ms) using the Neon transfection system. The knockout efficiency was monitored by measuring CDK1 expression using flow cytometry; it took 10–12 days after electroporation to detect decreased expression of CDK1.
RT–qPCR analysis
After cell sorting, DNA was extracted from 6–8 × 105 target T cells using DNeasy blood and tissue kit (Qiagen). Early RT and late RT products were quantified using primers for RU5 region (strong stop codon) and second-strand transfer, respectively57. 2-LTR circles were quantified using primers for the 2-LTR junction58. qPCR products were quantified using TaqMan Gene Expression Master Mix (Thermo Fisher Scientific) and the 7500 Real-Time PCR System (Applied Biosystems), using the following primers: RU5-forward: 5′-GCCTCAATAAAGCTTGCCTTGA; RU5-reverse: 5′-TGACTAAAAGGGTCTGAGGGATCT; RU5-probe: 5′-FAM-AGAGTCACACAACAGACG GGCACACACTA-TAMRA; second-strand transfer-forward: 5′-TAGTCAGTGTGGAAAATCTCTAGC; second-strand transfer-reverse: 5′-CTTCTAGCC TCCGCTAGTCAA; second-strand transfer-probe: 5′-FAM-TCGACGCAGGACTCGGCTTGCT-TAMRA; 2-LTR junction-forward: 5′-AACTAGAGATCCCTCAGACCCTTTT; 2-LTR junction-reverse: 5′-CTTGTCTTCGTTGGGAGTGAATT; 2-LTR junction-probe: 5′-FAM-CTAGAGATTTTCCACACTGAC-TAMRA.
HIV proviral integration was quantified from DNA extracts using the Alu-Gag PCR method as described previously16,59.
Quantification of cellular dNTPs
dNTPs were extracted from 8 × 105 cells per sample and quantified by qPCR using the single-nucleotide incorporation and PCR extension method as described previously60. To measure the effect of increasing intracellular dNTP levels, cells were incubated with exogenous deoxynucleosides (2 mM each of deoxyadenosine, deoxythymidine, deoxyguanosine and deoxycytidine; Thermo Fisher Scientific) as described previously61 for 24 h before HIV-1 infection.
Immunoblotting
For whole-cell lysates, cells (1.5 × 106 cells per sample) were washed in PBS and lysed in RIPA buffer supplemented with benzonase. Isolation of nuclei was performed as described previously62. In brief, 1.5 × 106 cells per sample were lysed in buffer A (320 mM sucrose, 10 mM HEPES, 8 mM MgCl2, 1× Roche EDTA-free cOmplete Protease Inhibitor) with 0.1% (v/v) Triton X-100 and nuclei were isolated by centrifugation. The nuclear fraction was washed twice in buffer A without Triton X-100 before lysis in RIPA buffer. Cell/nuclear lysates were separated by SDS–PAGE, transferred onto a nitrocellulose membrane and blocked in 5% skimmed milk, 0.05% Tween-20 in PBS or TRIS-buffered saline (for phospho-SAMHD1 detection) for 1 h. The blots were then probed with following primary antibodies overnight at 4 °C: TPR (PA5-54048, Invitrogen, 1:2,000), Nup153 (ab96462, Abcam, 1:2,000), Nup98 (C39A3, Cell Signaling Technology, 1:5,000), Nup62 (610497, BD), Nup54 (16232-1-AP, Proteintech, 1:5,000), Lamin A/C (ab108595, Abcam, 1:2,000) GAPDH (W17079A, BioLegend, 1:2,000), phospho-SAMHD1 Thr592 (D702M, Cell Signaling Technology, 1:2,000), total SMAHD1 (W19081C, BioLegend, 1:2,000) and tubulin (DM1A, Sigma-Aldrich, 1:2,000). After washing, blots were incubated with the following fluorescently tagged secondary antibodies for 1 h at room temperature (1:10,000): anti-rabbit IgG (RDye 800CW, Abcam), anti-rat IgG (Alexa680, Abcam) and anti-mouse IgG (IRDye 680RD, Abcam). Immunoblots were imaged using the Odyssey DXl Infrared Imager (LI-COR Biosciences) and analysed with Image Studio Lite software v5.2 (LI-COR Biosciences).
iSIM imaging
Resting primary T cells were VS-primed or bead primed as described above, infected with GFP–CA NL4.3 virus (1 U RT per 106 cells) for 6 h or 24 h and then transferred to an optical 96-well µ-plate (105 cells per well) (Ibidi) coated with poly-D-lysine (Thermo Fisher Scientific) for 2 h before being fixed with methanol-free 4% formaldehyde (Thermo Fisher Scientific) and washed in PBS. For KPNΒ1 localization assays, cells were treated with either DMSO or exportin-1 inhibitor for 3 h before fixation. For permeabilization, cells were incubated for 15 min with 0.25% Triton X-100 in PBS followed by washing in PBS. Cells were then blocked for 45 min at 4 °C with 3% BSA and 0.1% Triton X-100 in PBS followed by washing in PBS. Cells were incubated with primary antibody (1:100) overnight in 1% BSA in PBS at 4 °C, washed in PBS and incubated with secondary antibody (1:500) in 1% BSA in PBS for 1 h at 4 °C. The following primary antibodies were used: lamin A/C (ab108595, Abcam; sc-7292, Santa Cruz Biotechnology), KPNΒ1 (ab2811, Abcam), Nup358 (ab64276, Abcam), Nup214 (Abcam, ab70497), hCG1 (Abcam, ab192609), Nup54 (Proteintech, 16232-1-AP), Nup58 (Atlas Antibodies, HPA039360), Nup62 (SC48389, Santa Cruz Biotechnology), Nup98 (C39A3, Cell Signaling Technology), Nup153 (ab96462, Abcam), Nup50 (A301-783A, Fortis), TPR (ab58344, Abcam; PA5-54048, Invitrogen) and HIV-1 CA (ab309159, Abcam). The following secondary antibodies were used: anti-rabbit IgG (Alexa568, A-11011, Invitrogen) and anti-mouse IgG (Alexa568, A-11004, Invitrogen). Nuclear staining was carried out by incubating cells with NucSpot650 (Biotium) for 60 min at room temperature.
iSIM imaging was performed using a VT-iSIM imaging system (Visitech). Four-channel images (405 nm/488 nm/561 nm/640 nm) of the medial T cell plane were acquired using a CoolLED pE-4000 MultiLazer, Hamamatsu Orca camera and a Plan Apo ×60/1.4 NA oil objective with ×1.5 zoom. iSIM post-processing was applied in NIS-Elements (Nikon), using a five-iteration Richardson–Lucy deconvolution algorithm providing an effective xy resolution of 150–170 nm.
Live-cell iSIM imaging
Cells were incubated at 37 °C for 30 mins in Leibovits-L15 (no Phenol Red, Thermo Fisher Scientific) with 10% FCS with 1× CellBrite-Steady-550 (30107-T, Biotium) membrane stain and 1× NucSpot-650 (40082, Biotium) live nuclear stain. Cells were washed once in PBS and resuspended in Leibovits-L15 (no Phenol Red, Thermo Fisher Scientific) with 10% FCS, with uncoated 5-μm-diameter (Bangs Laboratories) biotin tagged BG505 SOSIP Env and seeded in an Ibidi µ-slide glass-bottom 18-well chamber (81817-900, Ibidi) precoated with poly-d-lysine (Thermo Fisher Scientific). Cells were transferred to environmental chamber (37 °C, 5% CO2) of the VT-iSIM imaging system (Visitech) and incubated for 1 h. The cells were then challenged with 2 U RT per 106 cells of HIV-1-StayGold-CA virus and imaging carried out immediately. Live-cell imaging was performed using the VT-iSIM imaging system (Visitech). Three-channel (488 nm/561 nm/640 nm), 21 slice (increment = 0.5 μm) z stacks were acquired using the CoolLED pE-4000 MultiLazer, Hamamatsu Orca camera and a Plan Apo ×100/1.45 NA oil objective. Continuous imaging was carried out for 4 h with acquisition intervals of 2 mins. In both conditions, cells were preselected for evident perinuclear HIV-1-StayGold virion localizations and cropped to single-cell fields of view for post-processing. iSIM post-processing was applied in NIS-Elements (Nikon), using a five-iteration Richardson–Lucy deconvolution algorithm providing an effective xy resolution of 130 nm.
iSIM image analysis
Resting target T cells were manually identified as cells labelled with eFluor450 Cell Proliferation Dye and the target population was collected by cropping a bounding box to the target T cell mask. Cropped target T cell populations were processed for quantitative analysis in Fiji imageJ63 using ImageJ macros (https://github.com/MattVXWhelan/Mesner_Whelan_et_al/blob/main/ImagejMacros). HIV-1 GFP–CA+ target T cells were manually scored for GFP–CA cellular localization using Fiji ImageJ’s plot profile function. GFP–CA+ puncta were classed as NE associated by colocalization of 50% of the puncta intensity histogram with perinuclear lamin A/C or TPR. Puncta were classified as nuclear by colocalization with NucSpot650 nuclear signal. Surface/cytosolic GFP puncta were determined as the remainder of the T-cell-associated GFP–CA+ puncta. Automated image analysis of Nuclear/NPC TPR and Nup62 was carried out using Cellprofiler 464. T cell nuclei were segmented from a mask generated from Nucspot650. Nucleoporin puncta were segmented and nucleoporin intensity was quantified within the nuclear mask which encompassed peripheral NPCs. Cell profiler pipelines are available online (https://github.com/MattVXWhelan/Mesner_Whelan_et_al/blob/main/CellProfiler_Analysis). Single-NPC quantification was carried out using the Fiji ImageJ analysis plot profile function to measure the pixel intensity of HIV-CA-associated Nup62/TPR-positive NPCs. For live-cell image analysis, pre-processing steps were applied in Fiji ImageJ. This included xyz drift correction using NucSpot650 channel as a guide using Fast4Dreg65, followed by ImageJ macros applying a 50 px rolling-ball background subtraction followed by autothreshold of channel intensities for visualization. HIV-SG puncta were identified through fitting of a Gaussian function (Fiji ImageJ, Thunderstorm plugin) and manually tracked in 3D. Perinuclear localization was visually scored by localization of HIV-1-StayGold full width at half maximum (FWHM) to the adjacent NucSpot650 signal. Nuclear localization was determined by peak intensity colocalization of HIV-1-StayGold virions with NucSpot650 Intensity. Live-cell ImageJ macros used in analysis are available online (https://github.com/MattVXWhelan/Mesner_Whelan_et_al/blob/main/LiveCellPipeline).
dSTORM imaging
The samples were prepared and analysed using the ONI Discovery kit (Oxford Nanoimaging). Nucleoporins were labelled with anti-FG-Nups antibody (MAb414, ab24609, Abcam, 1:100) and anti-Nup54 (16232-1-AP, Proteintech, 1:100) or anti-lamin A/C (ab108595, Abcam, 1:100). Primary labelled Nups were labelled with CF568-cojugated secondary antibodies (Biotium, 1:500) and Spot-tagged CA virus with Spot-Label Alexa Fluor 647 nanobody (Proteintech, 1:200). dSTORM imaging was carried out on the ONI Nanoimager (Oxford Nanoimaging) in the presence of ONI BCubed buffer. After bead xy channel calibration, two-channel images were acquired on HiLO or TIRF mode at 30 ms for 10,000 frames. Localization renderings were generated in NimOS software (Oxford Nanoimaging). Nanoimager results filtering based on the manufacturer’s recommendations: photon count (>300), localization precision: xy (5–25 nm), sigma xy (50–250 nm). For visualization, localizations were also rendered in Fiji ImageJ using the ThunderSTORM plugin66.
dSTORM analysis
NPC HIV-1 interactions were quantified using a similar approach to that previously described25. For translocation analysis, localization frequencies were quantified using the NimOS line Histogram tool (width: 3 px, bins: 40). Diameter calculations were carried out by a Gaussian fit of localization frequency distributions followed by a FWHM calculation for each histogram (Prism GraphPad). For Nup54 localization measurements. A 2D Gaussian fitting of localizations was carried out in Napari (v.0.5)67. The analysis pipeline is available online (https://github.com/MattVXWhelan/Mesner_Whelan_et_al/blob/main/STORM_Analysis).
MS analysis
Sample preparation
Resting CD4+ T cells (1–1.2 × 107 cells per condition, 4 independent PBMC donors) were pretreated with CDK1/2 inhibitor (BMS-265246, 2 μM, Cayman Chemicals) or DMSO control for 1 h and stimulated with beads (anti-CD4, Env, anti-CD3/CD28) for 6 h. Cells were collected by centrifugation and washed twice in PBS. Cell pellets were boiled in lysis buffer (5% SDS, 5 mm tris(2-carboxyethyl)phosphine, 10 mm chloroacetamide, 100 mm Tris, pH 8.5) for 10 min followed by micro-tip probe sonication (Q705 Sonicator from Fisherbrand) for 2 min with pulses of 1 s on and 1 s off at 50% amplitude. The protein concentration was estimated using the BCA assay (Thermo Fisher Scientific). Protein digestion was automated on the KingFisher APEX robot (Thermo Fisher Scientific) in 96-well format using a protocol described previously68 with minor modifications. The 96-well comb is stored in plate 1, the sample in plate 2 in a final concentration of 70% acetonitrile with magnetic MagReSyn Hydroxyl beads (ReSyn Biosciences) in a protein/bead ratio of 1:2. Wash solutions are in plates 3–5 (95% acetonitrile) and plates 6–7 (70% ethanol). Plate 8 contained 300 μl digestion solution of 100 mm Tris pH 8.5 and trypsin (Promega) in an enzyme:protein ratio of 1:100. The protein aggregation was carried out in two steps of 1 min mixing at medium mixing speed, followed by a 10 min pause each. The sequential washes were performed in 2.5 min and slow speed, without releasing the beads from the magnet. The digestion was set to 16 h at 37 °C with slow speed. Protease activity was quenched by acidification with trifluoroacetic acid (TFA) to a final pH of 2 and the resulting peptide mixture was purified on the OASIS HLB 96-well plate (Waters). Purified peptides were dried in a Savant DNA120 (Thermo Fisher Scientific) system and 5% of the 200 µg total amount was separated and used directly for liquid chromatography (LC)–MS for proteomics analysis, while the rest was used for phospho-enrichment.
Phosphopeptide enrichment
Phosphopeptide enrichment was performed on the KingFisher APEX robot (with the protocol described previously68) using the MagReSyn Zr-IMAC HP beads (ReSyn Biosciences). The robot layout was as follows: plate 1: the 96-well comb; plate 2: 40 µl of Zr-IMAC HP beads dissolved in 160 µl of the loading buffer (80% acetonitrile, 5% TFA and 0.1 M glycolic acid); plate 3: 500 µl of loading buffer; plate 4: and samples dissolved in 200 µl of loading buffer. Plates 5–7 were filled with 500 μl wash solutions: plate 5 contained the loading buffer, plate 6 contained 80% ACN (acetonitrile) with 1% TFA and plate 7 contained 10% ACN and 0.2% TFA. Plate 8 contained 200 μl 1% ammonia for elution. The beads were washed in loading buffer for 5 min at medium mixing speed, followed by binding of the phosphopeptides for 20 min at medium speed. The sequential washes were performed for 2 min at fast speed. Phosphopeptides were eluted in 10 min at medium mixing speed. Eluted phosphopeptides were acidified with 10% TFA to pH < 3 and purified in an OASIS HLB 96-well plate (Waters).
LC–MS
Dried peptides and phosphopeptides were dissolved in 0.5% TFA analysed using the Ultimate3000 high-performance liquid chromatography system coupled online to an Orbitrap Eclipse mass spectrometer (Thermo Fisher Scientific). Buffer A consisted of water acidified with 0.1% formic acid, while buffer B was 80% acetonitrile and 20% water with 0.1% formic acid. The peptides were first trapped for 1 min at 30 μl min−1 with 100% buffer A on a trap (0.3 mm by 5 mm with PepMap C18, 5 μm, 100 Å; Thermo Fisher Scientific); after trapping, the peptides were separated by a 50 cm µPAC Neo HPLC column (Thermo Fisher Scientific). The gradient was 3–35% B in 48 min at 750 nl min−1. Buffer B was then raised to 55% in 3 min and increased to 99% for the cleaning step. Peptides were ionized using a spray voltage of 2 kV and a capillary heated at 275 °C. The mass spectrometer was set to acquire full-scan MS spectra (350 to 1400 mass/charge ratio) at a mass resolution of 120,000 and an automated gain control (AGC) target value of 250% (RF lens of 40% and maximum injection time of 45 ms). For MS/MS fragmentation, we chose the DIA approach, but we used different settings for normal or phosphopeptides: for normal proteomics, the m/z range used was 361–1,033 m/z divided in 56 windows (12 m/z each with 1 Da overlap) with an AGC of 1,000% and resolution of 15,000. For phosphopeptides, we used 28 windows of 24 Th each with an overlap of 1 (m/z range from 472–1,143) and a resolution of 30,000. All raw files were transformed in .htrms format and analysed by Spectronaut v.18.5 with direct DIA analysis. We used the library generated automatically using Human reference proteome (20,420 sequences downloaded from UniProt) together with the MaxQuant contaminants list and standard settings: for normal peptides we used the BGS factory settings and, for the phospho files, we used the BGS phospho PTM workflow.
Data analysis
Abundance proteomics data were analysed using R (v.4.1.1). Batch correction was performed using ComBat from the sva package (v.3.42.0) to account for donor-specific effects. No imputation was performed and proteins that were not detected in at least two donors across at least 60% of experimental conditions were excluded. Differential expression analysis was performed using the limma package (v.3.50.1) with donor as a blocking factor. Benjamini–Hochberg correction was applied. Phosphoproteome data were analysed using Perseus v.2.1.6.069. Phosphosites that were undetected in two or more donors per condition were excluded and the remaining missing values were imputed from normal distribution for each sample to calculate fold changes and statistical significance. The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD062217.
Statistical analysis
Statistical tests were performed using Prism v.10 (GraphPad) and details of statistical tests used are indicated. Statistical tests for proteomics data were performed using R and Perseus as described above.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.