
Spondyloarthritis is a group of chronic inflammatory diseases that are typically seronegative and cause pain and stiffness in the spine and other joints. Key subtypes include axial spondylarthritis, psoriatic arthritis, and reactive arthritis. This review focuses on axial spondyloarthritis (axSpA), while highlighting findings from other spondyloarthritis subtypes to underscore the importance of investigating comparable mechanisms in axSpA. The primary involvement in axSpA is symmetric sacroiliitis, while the other subtypes focus on peripheral joints. This has made research in axSpA difficult due to limited access to spinal tissue and the reliance on extrapolating peripheral joint findings to the spine. Fortunately, there is a significant genetic component in the pathobiology of axSpA, with genome-wide association studies (GWASs)1 having identified the association between genes in the IL-23/IL-17A pathway and axSpA susceptibility. Indeed, targeting IL-17A has been identified as an effective therapy for axSpA, but targeting IL-23 was determined to be ineffective.
The physiological control of IL-23/IL-17A pathways involves the balance between IL-17A-producing cells, such as TH17 cells, which are pro-inflammatory, and Tregs, which are anti-inflammatory. During acute inflammation, IL-23-stimulated TH17 cells proliferate and skew the axis towards pro-inflammatory signals through IL-17A for effective pathogen clearance. After clearance, homeostasis should be restored through Treg suppression. Thus, these cells are important for the maintenance of self-tolerance and immune homeostasis. In the context of chronic inflammation, as found in axSpA patients, this anti-inflammatory signal is insufficient, and inflammation persists. FOXP3 is essential for maintaining Treg suppressive function and for preventing the transition towards pro-inflammatory phenotypes. Multiple axSpA-associated polymorphisms converge on pathways regulating Treg function, suggesting that inherited defects predispose Tregs to functional instability. Treg behavior is tissue-specific, and this functional instability may explain different extra-articular manifestations observed in axSpA. Thus, it is important to consider the Treg function during axSpA disease. Three key hypotheses have been proposed to identify the role of Tregs in the pathogenesis of axSpA: (1) as innocent bystanders or as accessories to a crime, either in (2) a passive or (3) active role. There is evidence that supports each of these hypotheses, making it important to highlight the development, stability, and function of Tregs in the context of autoimmunity and inflammation.
What are regulatory T cells and where do they come from?
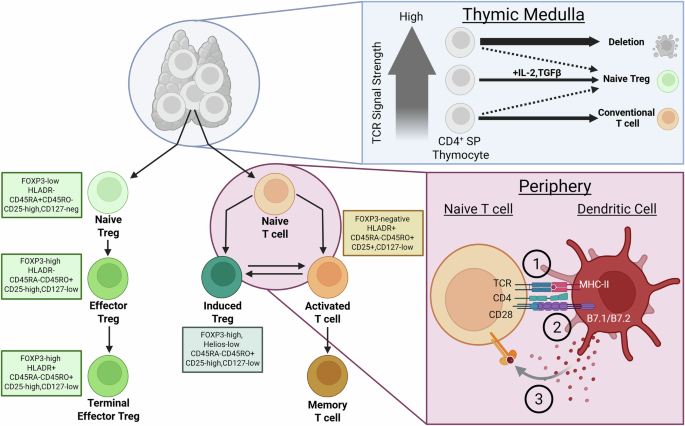
All T cell lineages have their origins in the thymus as thymocytes and later differentiate into either conventional or regulatory subsets after activation (see Fig. 1). The subset fate is determined through the T-cell receptor (TCR) signal strength and the presence of IL-2. A very strong TCR signal concurrent with IL-2 favors regulatory subset differentiation, and a weaker TCR signal without IL-2 favors conventional T cells2. This model of thymic-derived Tregs (tTregs) has been challenged though and TGFβ signaling has been suggested to play a greater role in the formation of tTregs. In BALB/c mice, the TGFβ signal protects tTregs from apoptosis and induces FOXP3 expression in CD4+ SP thymocytes3. These authors also demonstrate that although TCR affinity is important in tTreg generation, inhibition of TGFβ signaling drastically decreased FOXP3+ thymocytes. These thymic derived Tregs are vital to maintaining immune homeostasis and therefore are very stable to external pressures. Typically defects in these Tregs are quite severe and lead to debilitating disease early in life. This review will therefore focus on induced Tregs (iTregs) which derive in the periphery and are more responsive to external environmental signals. It is this subset that likely contributes to axSpA immune pathology in differing ways dependent on inflammatory environment.

Regulatory T cells (Tregs) in the thymus derive from CD4+ single positive T cells when the TCR signal is strong, and IL-2 + Transforming Growth Factor Beta (TGFβ) is supplemented. These Tregs are naïve to foreign antigen and therefore display a CD45RA+ phenotype. Upon antigen encounter, Tregs upregulate FOXP3 and CD45RO expression and are termed effector Tregs. In the periphery, a naïve CD4+T cell can become an Induced Treg or an activated T cell depending on the three signals received by an antigen-presenting cell. These three signals include (1) antigen presentation via MHC, (2) co-stimulation through B7.1/B7.2 and (3) cytokine signaling. CD4+ activated T cells can differentiate into an induced Treg based on the cytokine signal received in the microenvironment. Markers to distinguish Tregs based on development stage are listed in the box beside the cell. IL interleukin, SP single positive, CD cluster of differentiation, FOXP3 forkhead box P3, TCR T-cell receptor, HLA human leukocyte antigen, MHC major histocompatibility complex, B7.1/B7.2 CD80 and CD86. Figure was adapted from Sakaguchi et al. and Josefowicz et al.2,147 and created in BioRender. Pacheco, A. (2026) https://BioRender.com/012h2r5.
In the periphery, plasticity between CD4+ T cells means an activated effector T cell could differentiate into a Treg upon anti-inflammatory cytokine stimulation. This differentiation establishes immune homeostasis and avoids chronic inflammation from occurring following an infection. Interestingly, both TH17 and Tregs share the TGFβ signaling pathway in their differentiation. In the absence of IL-6 or IL-21, naïve CD4+ T cells differentiate into Treg cells4,5. These periphery-derived Tregs are generally less stable and can differentiate back into effector cells when stimulated with a pro-inflammatory signal. For example, synovial fibroblasts in autoimmune arthritis mouse models produce IL-6 to convert FOXP3+ Tregs into pathogenic TH17 cells6. In the presence of IL-6 and TGFβ and TCR stimulation, STAT3 is activated which induces the expression of the transcription factor RORγt, polarizing the CD4+ T cell towards the TH17 subset7. Tregs are not defenseless however and may develop stronger suppressive capabilities even if they gain the expression of RORγt when stimulated with pro-inflammatory stimuli. This subset of Tregs, known as effector Tregs, directs their suppressive capabilities in a manner specialized for the type of immune response activated.
Regulatory T cells in autoimmunity—importance of FOXP3
Plasticity is important for Tregs to adapt their phenotype and function based on changing environments. Through the regulation of environmental signals, Tregs can acquire features to control specific immune response types through the expression of master transcription factors (see Fig. 2). They therefore acquire T-bet expression to restrain type 1 inflammation8, Interferon Regulatory Factor 4 (IRF4) and GATA binding protein 3 (GATA3) for type 2 inflammation9, and STAT3 and RORγt for type 3 responses10. This review will focus on the Tregs important for suppressing type 1 and type 3 immune responses.

Regulatory T cells (Tregs), when activated to become an effector Treg, gain characteristics similar to the cell they are suppressing. The activation towards effector phenotype is dependent on the cytokine milieu stimulating the cell. Transforming growth factor beta (TGFβ) ensures Treg conversion while overstimulation from IL-12, IL-6, or IL-4 can lead to the conversion into T-helper 1 (TH1), T-helper 17 (TH17) and T-helper 2 (TH2) respectively. The conversion between T follicular regulatory (TFR) cells and T follicular helper cells (TFH) has not been demonstrated and is represented by a broken arrow. Suppression is displayed by the red arrow. IL interleukin, CXCR C-X-C motif chemokine receptor, Tbet T-box transcription factor 21, FOXP3 forkhead box P3, RORγt RAR-related orphan receptor gamma, GATA3 GATA binding protein 3, CCR C-C motif chemokine receptor 6, Bcl6 B-cell lymphoma 6 transcription factor. Figure created using BioRender. Tavasolian, F. (2026) https://BioRender.com/gp9pmm1.
RORγt and FOXP3 double-expressing Tregs represent one highly suppressive effector subset of Tregs in type 3 immunity11. In murine models, this subset shows transcriptomic and epigenetic signatures of Tregs and TH17 cells, but ultimately are more similar to Tregs since Treg-specific signature genes are demethylated12. RORγt+ Tregs express high levels of IL10, CTLA4 and ICOS, conferring superior suppressive capabilities on TH17 cells through cytokine-mediated and contact-dependent suppression12,13. Additionally, RORγt+ Tregs were shown to control intestinal inflammation in several colitis models12,14, and are significantly reduced in frequencies in germ-free mice14,15. The differentiation of murine RORyt+ Tregs is dependent on the transcription factor c-Musculoaponeurotic Fibrosarcoma Oncogene Homolog (c-MAF) which also plays a role in IL-10 secretion16. This highlights the importance in maintaining FOXP3 expression since loss of FOXP3 can lead to RORγt only being expressed on Tregs and therefore polarizing differentiation of CD4+ T cells to TH17 cells.
Expression of c-MAF is dependent on STAT3 activation17,18 and therefore responsiveness to IL-6. TGFβ, in synergy with IL-6, activates STAT3 signaling and downregulates FOXP3 through proteasome degradation19. Interestingly, STAT3 is necessary for IL-10 signaling and STAT3 expression by Tregs has been demonstrated to be critical for Treg-mediated suppression of TH17 cells20. Protein inhibitors that target activated STAT3 can reduce peripheral arthritis and gut inflammation, while rebalancing the TH17/Treg ratio in curdlan-injected SKG mice, a model of spondyloarthritis21. Recently a pharmacological inhibitor of glutamate oxaloacetate transaminase 1 was demonstrated to increase FOXP3 expression in TH17, therefore inducing the expression of RORyt+FOXP3+ iTregs22. Although the effect of this pharmacological inhibitor was replicated by another group, this group demonstrated the effect is likely due to an off-target effect downstream from GOT1 since GOT1 knockdown mice have increase TH17 cells and higher IL-17A production23. Whether the induction of RORγt+ Tregs through these therapeutically targets could improve symptoms in autoimmune patients remains unknown. Interestingly, IL-17A -secreting Tregs are still suppressive in vitro unless in the presence of high-dose IL-1β and IL-624. This suggests that this cell subset, especially if engineered to not express IL6R, may improve autoimmune disease while also secreting pro-inflammatory cytokines during acute infections. This suppressive behavior while producing pro-inflammatory cytokines contrasts TH1-like Tregs, which do not suppress effectively when producing IFNγ.
The balance between TH1 and Treg cell polarization is important for ensuring that acute inflammation remains transient and does not progress to a chronic state. Murine models have demonstrated that glutamine is important for tipping the balance between TH1 and Treg. Glutamine is a precursor to α-ketoglutarate, which is required for TH1 differentiation while inhibiting Treg differentiation in an mTORC1-dependent manner25. Cytokine stimuli can also skew the TH1-Treg axis. TH1 cultured in TGFβ before TCR stimulation has demonstrated the reprogramming of TH1 cells intro Tregs. This addition before TCR stimulation is critical to reduce mTORC activity, which biases towards effector cell phenotypes26. Alternatively, Tregs can gain TH1-like characteristics to control TH1 cell-based inflammation through the co-expression of FOXP3 and Tbet, driven by IL-12 signaling27. IFNγ-expressing Tregs are described but generally represent intermediates in the differentiation towards TH1 cell phenotype, which occurs once FOXP3 expression is lost28. IFNγ-producing Tregs display a demethylated FOXP3 locus, like a conventional Treg would29, but suppressive activity is dramatically decreased. Determining the difference between TH1-like Treg and IFNγ-producing Tregs is critical in differentiating a pathogenic versus a suppressive Treg.
The transcription factor IKZF1 has an important role in Tregs to prevent IFNγ production. IFZF1, in a complex with FOXP3 and IKZF3, binds to open chromatin to repress the production of pro-inflammatory genes, such as IFNγ30. When deficient, FOXP3 expression is destabilized and Treg suppressive function is impaired. Interestingly, there is a SNP near IKZF1 that associates with axSpA susceptibility (Table 1). This SNP was found to have a protective role in axSpA and was found to likely alter gene expression. It is possible that prevention of IFNγ production in Tregs plays an important role in maintaining homeostasis in patients, but the role this subset of cells may have on axSpA disease has not been studied, to our knowledge.
The transcription factor hypoxia-inducible factor 1α (HIF1α) promotes IFNγ production in Tregs. HIF1α induces the expression of genes required for glycolysis when the cell is in a low oxygen environment. HIF1α is positively regulated by PI3K-AKT-mTOR signals, which are dampened in Tregs. This dampened signal, therefore, prevents the induction of HIF1α expression in Tregs27. The deletion of von Hippel-Lindau (VHL), an E3 ubiquitin ligase that targets HIF1α, reduces FOXP3 expression and increases IFNγ production in Tregs, after HIF1α activity is increased31. Previous work has demonstrated that murine and human Tregs cultured with macrophage migration inhibitory factor (MIF) have increased IL-17 production and reduced suppressive capabilities32. The authors demonstrate that the frequency of IFNγ+ CD4+ T cells from the mouse spleen is reduced with MIF injection. Whether the frequency of IFNγ+ Tregs also changes with MIF exposure remains unknown. The same authors also later demonstrated that MIF interacts with HIF1A to drive SpA pathologies33. The two molecules were found to interact in most tissues and very evidently in neutrophils. Whether the molecules interact in axSpA lymphocytes remains unresolved, though this interaction should be explored further. It is possible that MIF and HIF mutually increase the expression of each other in Tregs, as observed in neutrophils, to drive reduced suppression and increased IFNγ production, especially in a hypoxic environment. The conversion of Tregs becoming inflammatory is an appropriate response to tumors or infections where hypoxia may occur, but in the context of chronic inflammation, this immune response could perpetuate inflammation.
Role of FOXP3 in regulatory T cell stability
FOXP3 is required for the generation of functional Tregs as it stabilizes the Treg signature gene expression profile34. Fate mapping experiments in mice have found that a large portion of FOXP3+ Tregs lose their FOXP3 expression and produce proinflammatory cytokines in the context of an inflamed microenvironment or when adoptively transferred into Recombination Activating Gene 2 (RAG2) −/− mice35. These ex-FOXP3 cells were then able to induce autoimmunity when adoptively transferred to Non-Obese Diabetic (NOD) mice36. Other studies show that only a minor population of FOXP3+ Tregs are unstable37, and most of the unstable FOXP3-expressing CD4+ T cells are actually activated effector cells that temporarily express FOXP3 and have low suppressive capabilities38. It is therefore important to consider what triggers and maintains FOXP3 transcription and the targets regulated by FOXP3 represented in mouse and human CHIPseq data39,40,41 (summarized in Fig. 3).

The transcriptional and epigenetic modifications that control Forkhead box P3 (FOXP3) expression, where blue arrows signify upregulation while red arrows signify repression of its target. Blue boxes mean the protein increases FOXP3 expression, while red boxes mean the protein downregulates FOXP3 expression. Transcriptional modifications that upregulate FOXP3 expression include: (i) Interleukin-2 (IL-2) binding to Cluster of Differentiation 25 (CD25) to activate Signal Transducer and Activator of Transcription 5 (STAT5)50. (ii) T-cell Receptor (TCR) activates a pathway consisting of Forkhead box O (FOXO), Nuclear Factor of Activated T cells (NFAT), and Activator Protein-1 (AP-1)42. This TCR signal can also lead to lower FOXP3 expression through activation of Mammalian Target of Rapamycin (mTOR) or complex consisting of Suppressor of Cytokine Signaling 3 (SOCS3) and Spi-B transcription factor (Spib). (iii) Transforming Growth Factor Beta (TGFβ) activates the SMAD-dependent pathway (SMAD2/3) directly or through the mitogen-activated protein kinase/extracellular signal-regulated kinase (MEK/ERK) pathway. SMAD-independent pathways are activated through TGFβ-activated kinase 1 (TAK1) or FOXO3/NFAT/AP-1 pathways43; (iv) C-C Motif Chemokine Ligand (CCL1) binds to C-C Motif Chemokine Receptor 8 (CCR8) to induce STAT3-dependent upregulation of FOXP3148. FOXP3 expression is epigenetically and post-translationally altered through ubiquitination, phosphorylation, and methylation, which lowers expression, and acetylation, which increases expression. IL-1β or lipopolysaccharide (LPS) binding to Toll-like Receptor 4 (TLR4) can activate STIP1 Homology and U-box Containing Protein 1 (Stub1), leading to lysine 48-linked FOXP3 ubiquitination55. Alternatively, IL-6 stimulation can decrease FOXP3 expression by increasing ubiquitination through hypoxia-inducible factor 1 alpha (HIF1α) activation56, or modulating chromatin binding by reversing TGFβ-induced FOXP3 acetylation149. Proviral Integration site in Moloney murine leukemia virus family kinase 1 and 2 (PIM1/2) can phosphorylate FOXP3 in the N-terminal to reduce protein expression and reduced suppression150. IL2 stimulation can also demethylate FOXP3 through upregulation of Tet methylcytosine dioxygenase 2 (TET2)151. Epigenetic and transcriptional modifications adapted from Qiu et al.152. Downstream from FOXP3, the transcription factor targets are grouped based on gene role in Treg suppression, reducing inflammation, and Treg stability. Red X over black arrow means FOXP3 represses the transcription of the target gene. FOXP3 target genes adapted from CHIPseq analysis40,41. NT5E 5’-Nucleotidase, PRDM1 PR domain zinc finger protein 1, FASL Fas ligand, CTLA4 Cytotoxic T-lymphocyte Antigen 4, GZM granzyme, GITR glucocorticoid-induced Tumor necrosis factor receptor-related protein, IFNγ interferon gamma, JAK2 janus kinase 2, PTPN22 Protein tyrosing phosphatase non-receptor type 22, PDE3B phosphodiesterase 2B, NRP1 neuropilin-1, IRF4 interferon regulatory factor 4, miR micro RNA, TNFRSF9 tumor necrosis factor receptor superfamily member 9. The figure was created in BioRender. Tavasolian, F. (2026) https://BioRender.com/fe0m2na.
Treg dysfunctions can be broken down into two axes: FOXP3 expression and global epigenetic signature. FOXP3 transcription is triggered by TCR stimulation through Activator Protein-1 (AP-1) and Nuclear Factor of Activated T cells (NFAT) binding its promoter42. Concurrent with TCR stimulation, TGFβ and IL-2 stimulation are required for FOXP3 expression. TGFβ stimulation activates the transcription factor SMAD3 and SMAD-independent pathways43, while IL-2 stimulation activates STAT5. Recently, axSpA patients were demonstrated to have reduced pSTAT5+ Tregs compared to healthy controls, and this subset increased with treatment44. This would suggest reduced FOXP3 expression due to inadequate response to IL-2. There is a SNP identified near IL-2RA that associates significantly with the risk of axSpA susceptibility, but this SNP appears to increase STAT5 phosphorylation45. This would suggest that the IL-2 signal received by Tregs may be outcompeted by other conventional cells. This would then influence FOXP3 methylation and contribute to reduced Treg stability.
FOXP3 has 3 conserved non-coding sequences (CNSs) regions that transcription factors bind to maintain FOXP3 stability46. Smad3 and retinoic acid bind to CNS1 (after TGFβ stimulation), while CNS2 is a Treg-specific demethylated region that is required for FOXP3 expression when Tregs divide47. This region is highly methylated in non-Treg cells and does not allow for the binding of ETS proto-oncogene 1 (Ets-1)48, CREB/Activating Transcription Factor (ATF)49, STAT550, or FOXP346. Tregs from active axSpA peripheral blood were found to have increased CpG island methylation in the CNS2 region of FOXP3 coupled with reduced FOXP3 mean fluorescence intensity51. This complements the study that found reduced pSTAT5+ Tregs and emphasized a dysfunction in IL-2 utilization by axSpA Tregs.
FOXP3 mRNA can also be regulated through microRNA and long noncoding RNA. Recently, the long noncoding RNA LOC645166 was found to be downregulated in T cells of axSpA patients. LOC645166 in Jurkat T cells was described to interact with miR-188-5p and NFкB inhibitor-D to regulate the expression of FOXP352. Reasonably, miR-188-5p expression was increased alongside reduced NFKBID expression in AS T cells from peripheral blood. A miR-188-5p inhibitor was able to enhance the expression of NFKBID and FOXP3 in Jurkat T cells. These findings provide a potential avenue for reduced Treg in axSpA, but the study does not describe concomitant treatment or peripheral involvement in the AS patients. Future directions should test this inhibitor in preclinical models to see if the increased FOXP3 expression translates to reduced arthritis.
FOXP3 is also regulated post-translationally through acetylation, phosphorylation, and ubiquitination. Acetylation of lysine residues provides FOXP3 stability and the avoidance of proteasomal degradation34. TGFβ is a potential inducer of FOXP3 acetylation, which enhances chromatin binding, while also demethylating the FOXP3 promoter53. Aryl hydrocarbon receptor (AhR) signaling has also been documented to increase acetylation of lysine residues of FOXP354. Contrarily, inflammatory stimuli, such as IL-1β or lipopolysaccharide (LPS), result in ubiquitinase STIP1 homology and U-box containing protein 1 (Stub1)-mediated FOXP3 degradation through Lysine 48-linked ubiquitination55. Stimulation with IL-6 and TCR also induces HIF-1α, leading to FOXP3 ubiquitination56. While these processes are fundamental to FOXP3 regulation and Treg stability, their roles in axSpA remain understudied and warrant focused investigation as a critical next step.
Function of regulatory T cells
Immune tolerance—prevention of autoinflammation
The molecular mechanisms of Treg suppression can be grouped into contact-dependent suppression and cytokine-mediated suppression (see Fig. 4). In the context of axSpA, human Tregs that express CD39 represent an interesting suppressive function due their ability to effectively control the induction of TH17-plasticity. Extracellular ATP is a danger signal that activates the inflammasome and creates an environment of strong inflammation. CD39 on Tregs can remove extracellular ATP from the environment and prevent TH17 generation57. CD39+ Tregs appear to be more effective in controlling TH17 since CD39− Tregs do not suppress IL-17 production. These CD39− Tregs also had a higher secretion of IL-17 when cultured alone and stimulated with PMA/ionomycin58. This suggests CD39 expression may support Treg stability while also being itself a mechanism of suppression. Interestingly CD39+ Tregs in axSpA demonstrate reduced expression of EZH2 and may be less stable than healthy control CD39+ Tregs44. This is only speculative though and an in vitro study comparing the kinetics of FOXP3 loss in axSpA Tregs compared to HC Tregs in the presence of inflammatory signals is necessary to support this hypothesis. Another study using single-cell transcriptome and cell surface protein analysis in 10 axSpA patients peripheral blood has revealed a decreased surface protein expression of CD39 and CD95 compared to 29 HCs59. Although this study can be extrapolated across populations due to the diversity of their recruitment, their findings need to be validated with a larger cohort. Additionally, patient characteristics such as biologic response or co-morbidities need to be further explored.

Regulatory T cells (Treg) suppress conventional T cells (Tconv) through different mechanisms, divided into contact-mediated or cytokine-mediated. All mechanisms demonstrate suppressive mechanisms validated in human Tregs, except for mechanisms that are in gray and not bolded. Contact-mediated suppression includes (i) Transforming growth factor beta (TGFβ) tethered to the surface of Tregs through Glycoprotein A Repetitions Predominant (GARP), which is activated upon physical contact with Tconvs63. (ii) CD95 can bind to CD95L on Tconv to induce apoptosis153. (iii) Cytotoxic T-lymphocyte Antigen 4 (CTLA-4) can bind to CD80 on Tconv cells to prevent co-stimulation or can bind to antigen-presenting cells to prevent CD28 from binding through trans-endocytosis130,154. (iv) LAG3 (lymphocyte activating 3) can inhibit major histocompatibility complex (MHC) II signaling through direct binding155. Cytokine-mediated suppression includes i) ATP hydrolysis to adenosine through CD3957. (ii) TGFβ signaling in an autocrine fashion to maintain Forkhead Box Protein P3 (FOXP3) expression62 or binding to Tconv to induce FOXP3 expression when co-stimulated with IL-261. (iii) IL-10 released by Tregs binds to IL-10 receptor (IL10R) on Tconv cells to convert to T-regulatory 1 cells (TR1)66 or binds to IL10R on dendritic cells to downregulate function. iv)Absorption of IL-2 through CD25 to prevent absorption by Tconv and limit proliferation60. (v) Apoptosis of Tconv cells through the release of Galactin-1156 or lysis of Tconv after releasing granzymes. (iv) Although human Tregs were found not to express IL-35 like murine Tregs157, IL-35 stimulation can enhance suppression by inducing conventional T cells to express IL-35158. IL, interleukin; CD, Cluster of Differentiation. The figure was created in BioRender. Pacheco, A. (2026) https://BioRender.com/xo88hoh.
Another interesting perspective in axSpA Treg biology is the suppressive mechanisms through cytokine environment changes. Tregs are able to change the cytokine environment through the absorption of IL-2 by CD25, and therefore inducing apoptosis of conventional T cells through cytokine deprivation60, or through the production of anti-inflammatory cytokines TGFβ, lingual antimicrobial peptide (LAP), and IL-10. TGFβ and LAP induce FOXP3 in conventional T cells61 and sustain the functional program of Tregs by maintaining the expression of FOXP3, CTLA4, and IL-10, while repressing the expression of RORγt62. TGFβ additionally can suppress effector T cells through cell-contact-mediated suppression, where tethered TGFβ on the surface of Tregs through GARP is activated once physical contact with effector T cells is initiated63. Once activated, TGFβ binds TGFβR1/2 on effector T cells and suppresses their proliferation64. Interestingly, TGFβ signaling appears altered in HLA-B27 rat models where T cells are hyperresponsive to TGFβ1 (measured by increased SMAD2/3 phosphorylation), but these cells have reduced TGFβ1 expression65. This highlights a potential mechanism for pathogenic TH17 differentiation in the B27tg rat, which could be extrapolated to SpA pathobiology. Whether a similar interaction occurs in axSpA remains to be studied, but this interaction between TGFβ receptors and HLA-B27 may contribute to both the inflammatory and new bone formation aspects of disease. Further, the role Tregs have in this interaction remains to be studied.
IL-10 converts conventional T cells to T-regulatory 1 (TR1) cells66, a cell subset that is FOXP3-negative but has regulatory functions during chronic antigen stimulation. TR1 are CD49b+ LAG3+ CD226+ CD4+ T cells that can kill target cells with greater efficiency than other cytotoxic CD4 T cell67,68. TR1 cells play an important role in controlling IL-23 production through LAG-3 suppression on CX3CR1+ macrophages and innate lymphoid cells type 369. This control is important since TR1 cells are responsive to IL-23 and respond through downregulation of IL-10 while maintaining IFNγ production70. IL-27 signaling contrarily induces Blimp-1-mediated IL-10 production in TR1 cells and can drive TH17 cells to secrete IL-10, demonstrating the ability of TH17 to lose pathogenicity and adopt a TR1-like phenotype71. The discrepancy between poor IL-23i responses and effective IL-17i therapy highlights TR1 cells as a particularly relevant subset to study in axSpA. Additionally, the cytolytic activity of TR1 cells has been demonstrated to occur against myeloid antigen-presenting cells in an MHC class 1-dependent manner. When MHC was blocked with a monoclonal antibody, granzyme and perforin expression in TR1 did not increase in vitro. The interaction of killer cell Ig-like receptors on CD4+ T cells with MHC class 1 has been proposed to be important for cytotoxicity in TR172. Since HLA-B27 confers the greatest risk for axSpA susceptibility, TR1 may have a greater role in the axSpA autoreactivity.
Autoreactivity—killing for the greater good
Cytolytic activity can either play a suppressive function or promote escape from regulatory responses. NKG2D+ CD4+ T cells have been shown to effectively kill Tregs through the interaction with NKG2DL on Tregs that releases cytotoxic molecules like perforin, GZMB, and FASL. This mechanism of cell killing has been demonstrated in systemic lupus erythematosus, leaving patients with reduced suppressive cells73. However, Tregs are not defenseless. Tregs can upregulate GZMB and the endogenous inhibitor serine protease inhibitor 6 to hinder Treg apoptosis and promote suppressive activity74. This cytotoxic activity is mediated by IL-2, stimulating Blimp-1 to increase GZMB production75. MHC class II-restricted cytotoxic T cells (ThCTLs) are CD4+ cytotoxic T lymphocytes that are highly differentiated effector cells and that kill in an antigen-specific matter76. When cytotoxic CD4+ T cells (ThCTLs) and Tregs are cocultured in the presence of IL-2, both cell populations upregulate cytolytic activity. At low concentrations of IL-2, Tregs become susceptible to ThCTL killing, but at high concentrations of IL-2, Tregs maintain Treg suppressive activity and induce apoptosis on ThCTLs77. This takeover of ThCTL in low IL-2 environments may be a last resort of the immune response to clear virus-infected cells by removing the anti-inflammatory signal provided by Tregs. When Tregs are depleted, the surplus of IL-2 in a tumor microenvironment of mice leads to the acquisition of cytotoxic activity by CD4+ T cells75. Alternatively, this acquisition may be important in efficiently clearing inflammation when Tregs become senescent.
Some researchers hypothesize that ThCTLs derive from Tregs as a last line of defense after Tregs or CD4+ T cells become terminally exhausted. In atherosclerosis-prone mice, exTregs and Tregs were sorted and sequenced to identify transcriptional profiles. The exTreg signature appeared to express cytotoxic genes, and these cells were able to kill cells in a CD107a degranulation assay78. The signature found in mice was projected in humans to be CD3+CD4+CD16+CD56+ cells, which were both inflammatory and cytotoxic. Using TCR sequencing, Treg CDR3 sequences appeared clonally expanded in this identified subset78. This demonstrates that this cytotoxic cell subset may derive from Tregs, but alternatively, the cell subset may just be recognizing similar antigens as the Treg. Considering cytotoxic CD4+ T cells increase in numbers and activity with age, while inducible Tregs appear to decrease with age, this transition towards a cytotoxic cellular profile from Tregs is certainly possible79. Cellular senescence occurs with aging, and cytotoxic CD4+ T cells are effective at clearing senescent cells. This transition towards cytotoxic CD4+ T cells may therefore be important in the effective clearing of senescent cells to avoid disease. The possible contribution of this pathway to diseases such as axSpA should certainly be an area of investigation.
Treg in different tissues—delineating axSpA disease manifestations through Treg biology
Tregs that reside in specific non-lymphoid tissue have been described and may guide the different extra-articular features observed in axSpA. Tregs at these non-lymphoid sites are described to be heterogeneous both transcriptionally and epigenetically, where only 9% of genes and 3% of open chromatin regions are shared across visceral adipose tissue, skeletal muscle, and intestinal Tregs80. Although this comparison was done in mice, this heterogeneity is assumed in humans, with studies demonstrating unique characteristics of Tregs localized in healthy or diseased tissues. This heterogeneity is therefore important to consider when linking Treg mechanisms to extra-articular features in axSpA or treatment responses. We therefore describe the current knowledge of Tregs in the skin, gut, and joints.
Tregs in the skin
The skin is an epithelial barrier that interfaces with external pathogens and toxins. Due to the microbiota being present through the skin, tolerance in this region is important to avoid chronic inflammatory triggering. Typically, skin Tregs are found in areas of high hair density in humans and represent 20% of adult skin-resident CD4+ T cells81. Although murine skin Tregs are transcriptionally similar to colonic Tregs, they uniquely express Dgat2 due to lipid synthesis in skin, integrin Itgae instead of Itga482, and Jag1, responsible for Notch signaling83. In humans, skin Tregs were also observed to express Arg284. Interestingly, Arg2 expression was reduced in psoriatic skin and could be further explored in SpA patients to observe whether this dictates the onset of the psoriasis rather than other extra-articular manifestations.
The gain-of-function variants in CARD14 and IKBKB are two SNPs that appear to greatly mediate psoriasis and related PsA immunopathology through the dysregulation of Treg homeostasis. Both variants increase NFкB signaling in murine αβ T cells, resulting in increased IL-23-IL-17 gene expression that coincides with psoriasis-like skin inflammation85. The IKBKB gain-of-function mutation was also found to increase IL-17-producing Tregs alongside psoriasis-like skin inflammation86. These Tregs retain suppressive function, but their IL-17 signal likely contributes to disease. When double mutation is present, the increased NFкB activation drives systemic inflammation and the features of PsA, suggesting that Treg dysregulation at both epithelial and axial sites contributes to broader tissue involvement. Although these variants have not been associated with axSpA susceptibility, it would be valuable to explore whether they help identify axSpA patients at risk of developing psoriasis. Future work should also determine whether other distinct NFкB-related variants, specifically those that associate with axSpA susceptibility, contribute to Treg-mediated pathology in axial disease.
Alternatively, the expression of respective integrin (ITGAE or ITGA4) could determine the co-morbidity present. Similar to intestinal Tregs, TGFβ signaling has a direct role in skin Treg-mediated suppression of inflammation. TGFβ signaling plays a critical role in maintaining the cutaneous barrier and promoting wound healing through retention of memory T cells, apoptosis of effector cells, and formation of Langerhans’ cells in the skin87. Additionally, there is evidence of reduced suppressive function and instability towards pro-inflammatory IL-17A+ cells. Although Tregs are increased in blood and lesional skin, they rapidly lose FOXP3 expression, and the frequency of IL-17A-producing cells increases in the presence of pro-inflammatory cytokines (IL-1β, IL-6, and IL-23)88. This supports the idea that similar characteristics of reduced stability and function are observed across tissue sites, and the determination of disease site may be due to an antigen present or preference of integrin expressed. These findings remain to be extended to axSpA patients.
Tregs in intestinal tissues
The intestine is another epithelial barrier that provides immune defence against microbial and viral pathogens, with the majority of immune cells within the mucosa or innermost layer of the intestinal wall. Intestinal Tregs are necessary to promote immune tolerance and avoid chronic inflammation and therefore represent 35% of residing CD4+ T cells in the lamina propria and 25% of residing CD4+ T cells in the intestinal mucosal wall81. Intestinal Tregs are considered very adaptable to their tissue environment and often express RORγt with low amounts of Helios, reflective of a peripheral-derived Treg catered towards TH17 immunity81. Interestingly, circulating RORγt+ Tregs were observed to be reduced in axSpA peripheral blood compared to healthy donors89. This reduction may contribute to the unregulated IL-17 signal observed in axSpA patients. Contrarily, this reduction, since it was observed in peripheral blood, may be due to increased trafficking to the site of inflammation.
The terminal ileum of axSpA patients, when absent of bowel inflammation, displays significant upregulation of IL2, TGFβ1, FOXP3, STAT5, and IL10 transcripts90. There is a 5-fold increase in the proportion of Treg cells in the AS gut compared to healthy donors, and the majority of these cells produce IL-10. This would suggest that our reduced RORγt+ Treg finding may be due to increased trafficking to the gut. The authors also found that while TGFβ expression correlates with FOXP3 in axSpA patients, a correlation that did not occur in Crohn’s Disease patients. This suggests that TGFβ supports different immune mechanisms across the two diseases, where it serves an anti-inflammatory role in axSpA gut. This further explains the reduced TH17 polarization in axSpA gut despite high levels of IL-23. If Tregs are supportive for axSpA patients in the gut microenvironment, it would be interesting to explore why Tregs are not preventing disease in the joint. Additionally, since some axSpA patients develop inflammatory bowel disease, it would be interesting to explore whether a Treg defect determines the onset of this comorbidity.
Tregs in the joint
The limited accessibility of the sacroiliac joint has made the study of this microenvironment reliant on the findings of peripheral joints during arthritic flares. Although inflammation contributes to these arthritic flares, subsets of effector-like Tregs have been described in the synovial fluid (SF) of axSpA patients91, implicating the importance of Tregs in joint inflammation. Simone et al. described 10 different Treg subsets found in the SF of axSpA and psoriatic arthritis patients. They reported one subset, KLRB1+ Tregs, as an important suppressor of TNF and IL23 produced by monocytes. This Treg subset highly expressed LAG3, and LAG3 fusion proteins inhibited sorted monocytes production of TNF and IL2391. It is worth noting that direct suppression assays of these different subsets were not performed and the precise effect these Treg subsets have on joint inflammation remains unclear. The persistence of inflammation even with the presence of these suppressive Tregs remains unanswered.
While Tregs in axSpA patients appear to operate in the gut, another group explored potential reasons why Tregs fail to dampen joint inflammation. In TNFΔARE mice models, TNFR2 (TNFRSF1B) expression levels on Tregs are higher in the ileum compared to the synovium when under chronic TNF exposure92. Since TNFR2 typically activates regulatory mechanisms, this data suggests that Tregs may have a greater role in suppressing gut inflammation rather than joint inflammation under chronic TNF exposure. The authors also later suggest that under chronic TNF exposure, Tregs in the joint have increased expression of genes involved in canonical pathways such as apoptosis and oxidative stress compared to Tregs in the gut. The driver of these DEGs was hypothesized due to altered TNFR2 signaling, but the reason for reduced TNFR2 (TNFRSF1B) expression in the joint remains unclear. Additionally, although a similar difference between gut and joint Tregs may be observed in human SpA, future studies should examine synovial tissue and gut samples from SpA patients to determine if a similar site-specific Treg suppressive capability is observed. A comparison between spinal joint and peripheral joint may also demonstrate key Treg behavior differences and may better delineate axSpA from other more peripheral-dominant SpA diseases.
The role Tregs have in the joint may also further differ based on tissue site. A single-cell RNA sequencing atlas of human entheseal tissue was recently described from patients undergoing elective spinal surgery. Entheseal Tregs demonstrated higher expression of TGFβ1 but lower frequency compared to healthy ileum93. Interestingly, enthesis-derived mesenchymal stem cells (MSCs) were found to suppress T cell activation through the CD39/CD73 ectonucleotidase axis, highlighting an alternative immunoregulatory mechanism that may reduce the reliance on Tregs. This study was limited to healthy control tissue, however, and Treg migration in the enthesis may occur during SpA flares. Although the authors compare healthy enthesis to healthy ileum, the enthesis is generally sterile and may require fewer Tregs than the ileum to reduce inflammation in the healthy state. A key future direction will be to characterize Treg function in the diseased enthesis and to determine whether MSC-mediated regulation remains dominant. It also remains unknown whether axSpA entheseal MSCs mirror the reduced CD39 expression observed in peripheral blood Tregs, representing an additional important future study.
Although Tregs may play a less prominent role in entheseal tissue, they may have a significant role in bone repair and regeneration. In axSpA, new bone formation and bone loss can occur concurrently. Tregs are key players in bone healing and regeneration by suppressing osteoclastogenesis through inhibition of receptor activator of nuclear factor kappa-B ligand (RANKL) signaling and by contributing to osteoblast differentiation and supporting bone formation through TGFβ production. Tregs also support MSC-based bone repair by suppressing CD4 + T cells that produce IFNγ and TNFα94. While pathologic TH17 that produce IFNγ and IL-17A have been described to have a role in joint inflammation95,96, the reasons why Tregs fail to fully suppress this inflammatory axis in axSpA remain unclear. One possibility is that chronic inflammation initiates joint disease with inflammatory damage, whereas increased Treg may facilitate new bone formation. Consistent with this, axSpA patients with new bone formation exhibit a higher ratio of Tregs:TH17 compared to those without, but these findings remain correlative97. Additionally, increased IL10 mRNA in axSpA Tregs has been correlated with new bone formation97 Functional studies are necessary to determine if Tregs actively promote ankylosis in late disease, represent compensatory bystanders, or are functionally impaired in ways that permit pathogenic effector responses to persist.
Evidence for three unifying hypotheses for regulatory T cells in axial spondyloarthritis
The role of Tregs in axSpA pathobiology is not well defined yet and therefore requires further study. The frequency of Tregs in axSpA peripheral blood (with and without biologic treatment) is highly debated and is dependent on how the Tregs are defined (Table 2)98. These discrepancies likely reflect a combination of methodological differences (gating strategy, sample source) and biological variability (patient treatment status, disease duration, and activity). The function of Tregs in axSpA remains unresolved but can have a role in three different ways (Fig. 5). Tregs in axSpA could be (1) losing the war against inflammatory cells (Innocent bystanders) (2) losing suppressive capabilities and gaining inflammatory properties (Good cells gone bad) or (3) overcontributing to autoimmunity (Bad from the start).

Regulatory T cells (Treg) may have a role in axial spondyloarthritis (axSpA) in three different ways. They may transition towards pro-inflammatory cells such as T-helper 17 (TH17), T-helper 1 (TH1), or T-helper 1.17 (TH1.17) due to a lack of stability and therefore contribute to autoinflammation. They could end up producing proinflammatory cytokines such as interleukin 17A (IL-17A) or interferon gamma (IFNγ). Alternatively, they can gain exhaustive phenotypes and lose the capability to control inflammation. Tregs could convert to cytotoxic T cells and contribute to autoimmunity in a major histocompatibility complex class 2 (MHC-2) manner or gain T regulatory 1 (TR1) cell-like phenotypes and contribute through MHC-1. In both scenarios, high amounts of perforin (PFN), IFNγ, tumor necrosis factor alpha (TNFα), and granzymes (GZM) would be released to induce cell death. Lastly, Tregs could be weakened by cytokines such as interferon alpha (IFNα) or exosomal signaling, leading to an imbalance between regulatory and effector cells. Figure created in BioRender. Lim, M. (2026) https://BioRender.com/wsw56ci.
Losing the war against inflammation (
Innocent Bystanders
)
The Innocent Bystanders hypothesis would suggest that Tregs are developing normally, resistant to pro-inflammatory differentiation, and are functionally suppressive. This hypothesis would best be supported through a suppressive assay demonstrating that healthy control and axSpA patients can both suppress healthy control conventional cells, but both poorly suppress axSpA conventional cells. Although this has not been formally studied, there is evidence that axSpA Tregs are overwhelmed by the inflammatory signals but remain functionally important for patient improvement. The TH17/Treg balance was found skewed towards TH17 in axSpA patients compared to healthy controls, and this balance was restored with TNFα inhibition where Treg frequency inversely correlated with serum CRP levels99. This suggests Tregs have a role in reducing inflammatory markers in axSpA, but the overactive pro-inflammatory signal by TH17 overwhelms Treg capacity, where external biologics are required to obtain homeostasis. A study including 222 axSpA patients during TNFα inhibitor therapy supports this theory since it demonstrated an increase in Tregs after 6 months of treatment, but only in patients that demonstrated a clinical response. The change in frequency of Tregs was accompanied by decreased serum levels of TNFα, IL-6, IL-17 and IL-23, and an increase in TGFβ concentrations. Patients who did not respond to TNFi therapy demonstrated a significant increase in IL-17 and IL-23 and a decrease in Treg frequency100. It remains uncertain whether Tregs are the causative drivers of clinical improvement or simply downstream consequences of reduced systemic inflammation. The difference between patient therapeutic response does highlight the importance of Tregs in maintaining the reduced inflammatory signal achieved with biologic therapies. However, this phenomenon has not been observed in every study. One study found that TNFi treatment resulted in a decrease in Treg frequency, which was positively correlated to BASDAI and the serum levels of proinflammatory cytokines. This positive correlation was postulated to be a failed attempt by the patient’s immune system to reduce the proinflammatory response before biologic treatment was provided101. It is important to note that treatment was recorded only after 3 months, and the patient’s clinical response was not stratified in the analyses. A longitudinal study that measures Treg dynamics using lineage tracing in parallel to clinical endpoints would best distinguish whether Tregs have a causational role in clinical improvement or simply improve in response to reduced systemic inflammation.
Genes activated in response to interferon stimulus also appear to be more pronounced in Tregs from synovial fluid compared to peripheral blood, thus highlighting the potential of interferons driving Treg phenotype in an inflamed environment91. IFNα inhibits suppressive function against conventional T cells and NK cells by disturbing TCR signaling in Tregs through repression of cAMP102 and limiting the release of IL-2 by the Tregs103. Interestingly, CD4+ T cells cultured with exosomes from axSpA patients induce the production of IFNα and reduce the number of IRF4+ Tregs104. This external signal may therefore turn off Treg effector capabilities in axSpA patients and drive inflammation. Type 1 IFNs have recently been described to have a role in TNF inhibitor and IL17A inhibitor treatment refractory patients. The role type 1 IFNs have on Tregs should therefore be further studied to determine if reduced Treg function is driving treatment resistance.
The proinflammatory cytokine MIF is another signal that may reduce Treg capabilities. In human and mouse Tregs, MIF was shown to drive the acquisition of TH17-like phenotypes in vitro32. We recently identified a reduced frequency of RORγt+ Tregs in axSpA patient blood compared to age-matched healthy donors89. This Treg subset suppresses type 3 immunity most effectively and should be elevated in axSpA patients to reduce IL-17A signals. This may suggest that this subset is losing FOXP3 expression due to increased inflammatory pressures like MIF. Alternatively environmental factors may be driving this reduced frequency and allowing proinflammatory signals to drive further Treg conversion. RORγt+ Tregs are important in the gut and lymph nodes to prevent the occurrence of food intolerance and inflammatory bowel disease. The gut microbiome has been found to influence this Treg subset expansion through the release of tryptophan. Tryptophan deficiency increases the frequency of RORγt+ Tregs while AhR ligand indole-3-carbinol restores Treg homeostasis105. An altered tryptophan metabolism has been identified in axSpA, with some studies showing a relationship with clinical activity106. Mice with reduced RORγt+ dendritic cells also had reduced RORγt+ Tregs and impaired tolerance to oral antigens. An altered microbiome may therefore predispose patients to develop axSpA symptoms once they are exposed to a particular food antigen. A reduction of RORγt+ Tregs due to dysregulated training by commensal bacteria in early life may contribute to flares observed in later stages of disease. Beyond these findings, evidence of Treg plasticity in human axSpA patients remains limited. TCR repertoire studies of CD4+ T cells are necessary to determine whether the effector cell conversion of Tregs occurs in axSpA pathobiology. It remains evident from these findings that the study of transcription factors that regulate FOXP3 expression is crucial in the context of axSpA.
Loss of suppressive capabilities, gain of inflammatory properties (
Good Cells Gone Bad
)
The Good Cells Gone Bad hypothesis suggests axSpA Tregs have fundamental irregularities in stability or function. There indeed are identified genes associated with axSpA susceptibility that hint at Treg dysfunction. These single-nucleotide polymorphisms (SNPs) are summarized in Table 1.
The function of Tregs may be disrupted due to the SNPs associated with axSpA susceptibility. The rs2236279 missense polymorphism identified in axSpA and Crohn’s disease is in exon 9 of PRKCQ, a gene that encodes the protein kinase C-theta (PKC-θ). Being a missense polymorphism, protein activation and interactions may be altered, but the exact function of this SNP remains to be studied. PKC-θ inhibition enhances Treg function, protects Tregs from TNFα inactivation, and is found to restore Treg activity in defective Tregs from rheumatoid arthritis patients107. This SNP may therefore be common across Crohn’s disease and axSpA patients due to increased activity of PKC-θ and possibly reduced protection of Tregs from TNFα inactivation. An interesting study would therefore be to observe FOXP3+ Treg stability and activity in the gut and joint of murine models that have the rs223679 polymorphism.
The stability of Tregs may be altered by genetic polymorphisms in the patient. For example, the gain-of-function SNP rs4077515 in CARD9 is demonstrated to protect CK2-mediated depletion of CARD9, which may reduce Treg differentiation and promote TH17108. Although the increased TH17 expansion has been described by neutrophil mediation in SKG mice and in vitro neutrophil-TH17 co-culture109, the role this SNP has on Treg differentiation remains unknown. An interesting study to explore is whether the CARD9 gain-of-function changes the kinetics of Treg conversion to TH17 when co-cultured with neutrophils. A recent study describes how neutrophils can impede Treg differentiation. Neutrophil extracellular traps are described to be increased in spine enthesis tissues and SKG enthesis110. When cultured with CD4 + T cells, the differentiation into Tregs was reduced through the activation of TLR7 signaling. It would be interesting to observe if AS patients with the CARD9 gain-of-function have increased neutrophil extracellular traps and if this correlates with Treg frequency.
Reduced stability in axSpA Tregs is suggested since axSpA peripheral blood Tregs demonstrate lower FOXP3 mean fluorescence intensity than healthy controls and greater methylation in the CNS2 region of FOXP3. This is likely due to decreased phosphorylation of STAT5 and represents an effect downstream from these Tregs inability to uptake IL-251. Interestingly, there are polymorphisms near the IL-2 receptor that are associated with axSpA. These are found to increase IL-2RA expression and increase STAT5 phosphorylation in effector T cells and Tregs through45. Since these cells compete, it is possible that this polymorphism is more beneficial to effector T cells, allowing them to respond to IL-2 doses that typically only activate Tregs. Alternatively, the reduced phosphorylation in Tregs may be due to interactions with other proteins. Reduced STAT5 phosphorylation was also found in a separate cohort and this was coupled with decreased EZH2 expression, another marker of Treg stability44. This would suggest that the effector T cells are hyperproliferative while the Tregs are being deactivated through alternative mechanisms. IL-2 supplementation would therefore benefit effector T cells until these alternative mechanisms that are deactivating the Tregs are turned off. These alternative mechanisms remain to be determined but the study of the post-translational modifiers of EZH2 in axSpA is a reasonable next direction for determining potential drug targets to restore Treg function.
The identified SNPs associated with axSpA susceptibility, especially those with predicted or validated evidence, provide evidence that axSpA Tregs are weakened due to inherited genetic factors and shift towards pro-inflammatory CD4 + T cells in axSpA patients. After validation through experimentation, this hypothesis should be extended to determining the role Treg defects may have in treatment response vs. non-response. The additional SNPs may provide threats to Treg integrity and therefore reduce the ability for an anti-inflammatory response to develop when external treatment is supplemented. The patient would therefore require greater amounts of treatment and rely on cycling or switching of multiple treatments. This may also suggest diminished treatment effect, especially with increased inflammatory burden due to environmental factors. A future study could sequence Treg SNPs and correlate the number of hits with lines of therapy required to reach remission. Alternatively, the treatment type may just be incorrect for the patient and knowledge on their polymorphisms may guide correct treatment response.
Direct contributors to autoimmunity (Bad Actors from The Start)
The Bad Actors from the Start hypothesis suggests axSpA Tregs have developed normally and remain regulatory, but their suppressive capabilities via cytotoxicity are overproductive. This would suggest that axSpA Tregs contribute to increased TR1s through IL-10 induction of conventional T cells or by becoming terminally exhausted and remaining in a ThCTL state. Although both hypotheses require further investigation, there is evidence that suggests this theory should be explored further.
Bioinformatic analysis revealed cytotoxic markers (GZMA, GZMK, PRF1, GNLY, NKG7, KLRB1, KLRD1) could potentially be a biomarker for axSpA detection111. Although this finding implicates CD8+ T cells and NK cells in disease manifestation, the overlooked role of CD4+ T cells in altered cytotoxicity remains important. We recently described a reduced cytotoxic CD4+ T cell in a subset of axSpA patients, segregated based on biologic response89. Although patients who respond to anti-IL17A (secukinumab) treatment have elevated granzyme A expression in CD45RO+ CD4+ T cells before and after treatment, the expression never returns to normal in patients unresponsive to secukinumab treatment. Cytotoxicity in CD4+ T cells is important for killing tumor or virus-infected cells, but overproduction or misdirected targeting can contribute to autoimmunity. A subset of CD28− CD4+ T cells with the ability to produce IFNγ and perforin has been described in patients with axSpA112. This subset has been described to express TLR4 and TLR2 to a higher degree than other cell subsets, and this expression can be increased when cells are cultured with TNFα. When stimulated with LPS, these cells produced cytotoxic cytokines such as perforin113. This cell type had a higher ratio of CXCR3 to CCR4, produced more IFNγ, TNFα, and IL-10 in comparison to CD28+ cells, but lacked IL-2 and IL-4 production114. This provides a potential link between infectious agents and cytotoxic CD4+ T cells. Alternatively, this cell subset also expressed CD57 and other MHC class 1 recognizing receptors112. This implies that the cytotoxic CD4+ T cell may be involved in an HLA-B27-mediated mechanism that drives axSpA pathobiology.
TR1 cells are the other cytotoxic CD4+ T cell population that can mediate both regulatory and cytotoxic effects in response to MHC class 1 signaling. Interestingly, axSpA patients’ cytokine environment appears to be suitable for the expansion of TR1 cells115. Although the role of this cell subset in axSpA is unclear, ERAP −/− mice appear to have reduced numbers of TR1 cells. This knockout may be protective to axSpA indicating a reduced number of TR1 cells may actually improve axSpA symptoms116. Although a regulatory cell subset, incorrect antigen processing, aberrant cytotoxicity, or uncontrolled regulation of the subset from becoming pathogenic may explain why the removal of a regulatory cell subset could improve disease. Regardless, it is important to define TR1 cells’ role in axSpA and evaluate the cell’s role during biologic therapy to provide insight into novel therapeutic targets and strategies.
Forward-looking outlook
These three hypotheses do not represent strictly mutually exclusive scenarios but rather could provide a unifying framework that highlights phases along a dynamic continuum of Treg dysfunction in axSpA. We propose a model that highlights a dynamic trajectory of Treg dysfunction that underlies disease severity. In this model, early disease is characterized by intact Tregs as innocent bystanders become overwhelmed by excessive pro-inflammatory cues. As inflammation persists, chronic cytokine exposure, metabolic stress, and epigenetic instability erode FOXP3 expression, tipping Tregs into a dysfunctional state, (Good Cells Gone Bad). This stage is marked by reduced suppression and acquisition of pro-inflammatory phenotypes (IL-17A or IFNγ production). In parallel, or subsequently, certain regulatory subsets such as TR1 cells may acquire excessive cytotoxicity which perpetuate tissue damage and immune dysregulation. This model emphasizes that Treg dysfunction in axSpA represents a continuum that is temporally and contextually regulated, driven by the interplay of genetic predisposition, environmental triggers, and the local inflammatory milieu.
This model generates a testable hypothesis in axSpA which can advance our understanding of the breakdown of immune regulation and provide novel therapies based on disease stage. Current mechanistic understanding of Treg stability, plasticity and function in axSpA relies disproportionately on preclinical murine models of autoimmunity and inflammation, but advancements of experimental tools now allow for translation to human Treg. Although access to tissue samples has been a major challenge in studying axSpA, there have been recent advancements in enthesial and spinal tissue extraction. Spinous processes entheseal soft tissue and peri-entheseal bone have been extracted from non-spondyloarthritis patients during elective orthopedic procedures117. Although extrapolated to SpA patient synovial fluid and blood samples, there is an opportunity to complete this similar methodology on surgical samples from SpA patients to confirm if the findings could indeed be extrapolated. Achilles tendon enthesis needle biopsy has been completed118 and recently has been extended to axSpA patients119, and psoriatic arthritis patients during secukinumab treatment120. These studies employed histology, imaging mass cytometry and spatial transcriptomics to measure the cellular components in the biopsy and their changes during therapy. Although a Treg cluster is identified using spatial transcriptomics, the cluster is not further characterized. When the opportunity presents, immune profiling of Tregs using CyTOF, spatial transcriptomics and multi-omics integration would better delineate Treg heterogeneity across different disease states and tissue sites. A high parametric immune profiling panel across the different extracted tissues could explain the different results observed across different research groups due to heterogeneous methodologies summarized in Table 1. Additionally, high-parameter approaches are particularly advantageous when only limited cell numbers are available for analysis, a critical consideration given the restricted accessibility of axSpA tissues.
A longitudinal profiling of axSpA patients during the progression of disease could determine whether Treg plasticity toward pro-inflammatory states correlates with disease progression or treatment response. Recent technological developments, such as single-cell RNA-seq, ATAC-seq and TCR clonotyping, can now delineate the transcriptional and epigenetic landscape of Tregs across disease stages and treatment responses. These predicted trajectories can functionally be tested in vitro using defined cytokine conditions that mirror the inflamed joint environment. The creation of therapies that address the different stages of disease can then be predicted.
A therapy that targets metabolic pathways (like mTOR inhibitors) or stabilizes FOXP3 expression (such as a STAT5 agonist) might prevent the transition from overwhelmed Tregs towards dysfunctional Tregs. Such a therapy might be less effective in later disease stages, however, once Tregs have transitioned to a dysfunctional phenotype. In a later disease stage, Treg suppression and stability may be completely lost, necessitating the supplementation of new regulatory cells either through ex vivo expansion or by introducing specific extracellular signals to promote stable Treg expansion. Currently, there are 50 Treg cell therapy clinical trials listed on clinicaltrials.gov for the use to treat autoimmune disease and transplant rejection121. No drug-related serious adverse events have been reported in these trials; however, the progress is still preliminary, with most having only completed Phase 1 trials. Alternatively, an external cytokine or exosome signal could promote Treg differentiation to replace dysfunctional Tregs. A mesenchymal stem cell-derived exosome was recently described to promote Treg differentiation through the FOXP1/STAT5 signaling in a pre-clinical mouse model122. Although these findings represent a promising therapeutic approach, how long these newly differentiated Tregs exist and how well they suppress compared to pre-treatment Tregs remains unclear. Lastly, extracellular vesicles derived from Tregs represent a potential immunomodulatory therapeutic resistant to pro-inflammatory external signal influences123. This approach, like Treg cell therapy, would best address axSpA patients with a dysfunctional Treg phenotype. Although a promising approach, most applications are still in preclinical models, and insufficient evidence has been produced to yet allow progression towards human clinical studies.