
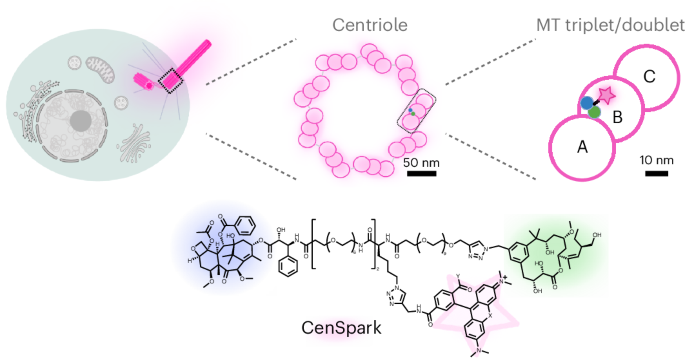
CenSpark probe generation and fluorescence excitation and emission measurements
The synthesis of CenSpark probes is reported in detail in Supplementary Information. CenSpark-650 and CenSpark-555 were kept in DMSO at −20 °C; SPY650-tubulin was obtained from Spirochrome. For fluorescence excitation and emission measurements, probes from the DMSO stock solutions were added to solutions containing tubulin (2 mg ml−1; Cytoskeleton, HTS03), 0.2% SDS (Applichem, A1502) or PBS (Thermo Fisher Scientific, 10010023). For the tubulin condition, 80 mM PIPES (Sigma-Aldrich, P6757), 2 mM MgCl2, 0.5 mM EGTA pH 6.9, 1 mM GTP (Cytoskeleton, BST06) and 15% glycerol were used. Samples were prepared in 1.5-ml microfuge tubes and incubated for 2–3 h at 37 °C; fluorescence was measured in a half-area 96-well plate (Greiner Bio-One, 675076) on a TecanSpark multimode microplate reader. For CenSpark-650, fluorescence excitation was determined by exciting from 450 nm to 690 nm, measuring emission at 720 nm, while fluorescence emission was determined by exciting at 550 nm, measuring emission from 580 nm to 850. For CenSpark-555, fluorescence excitation was determined by exciting from 350 to 590 nm, measuring emission at 610 nm, while fluorescence emission was determined by exciting at 430 nm, measuring emission from 465 nm to 750 nm. The excitation and emission bandwidths for all measurements were set to 10 nm. Excitation and emission spectra were normalized to the maximum value set at 1.
Cell culture and live imaging
RPE-1 (American Type Culture Collection, CRL-4000, lot 64139213) and RPE-1::tp53−/−::centrin 2–GFP50 cells were cultured in DMEM/F-12 (Thermo Fisher Scientific, 31331093) with 10% FBS (Merck, S0615), 0.2 mM sodium pyruvate (Thermo Fisher Scientific, 11360070) and MEM nonessential amino acids (Thermo Fisher Scientific, 11140-050). HeLa::centrin 1–GFP51 and U-2 OS (Sigma, 2022711) cells were cultured in DMEM (Thermo Fisher Scientific, 0565018) with 10% FBS. Cells were cultured in a humidified 5% CO2 incubator at 37 °C and split every 3–4 days or at confluency. Cells were seeded on glass-bottom µ-Dish 35-mm Quad dishes (Ibidi, 80416), 96-well glass-bottom dishes (Ibidi, 89627), µ-slide eight-well plate dishes (Ibidi, 80807) or 12-mm coverslips 1–2 days before imaging. Cells were seeded at 80% confluency when imaged for a single time point or at 10–15% confluency when imaged for 2 days in time-lapse experiments. The imaging medium contained Fluorobrite DMEM (Thermo Fisher Scientific, A1896701), GlutaMAX (100×, 1:100; Thermo Fisher Scientific, 35050061), 10% FBS and penicillin–streptomycin (100 U per ml; Merck, P0781) was added for long-term imaging experiments.
Microtubule doublet assembly
Unlabeled tubulin (T238P-B), biotin tubulin (T333P-A), HiL488 tubulin (TL488M-A) and rhodamine tubulin (TL590M-A) were from Cytoskeleton. GMPCPP (NU-405S) was from Jena BioScience. Subtilisin A from Bacillus licheniformis (P5380, referred to elsewhere as subtilisin), PMSF (93482), D-(+)-glucose (G8270), glucose oxidase from Aspergillus niger (G7141), k-casein from bovine milk (C0406) and methyl cellulose (M0512) were from Sigma-Aldrich. Microscope slides (10.0120.06) and coverglasses (22 × 22 mm; 10.0360.08) were from HuberLab.
First, 5 μl of a rhodamine-labeled tubulin mixture (68% unlabeled tubulin, 12% biotin tubulin and 20% rhodamine tubulin; 4 mg ml−1 overall) were diluted in MRB80 (80 mM PIPES, 1 mM EGTA and 4 mM MgCl2, pH 6.8) supplemented with 2.7 mM GMPCPP and preincubated for 5 min on ice. Microtubules were polymerized at 37 °C for 45 min and centrifuged at 150,000g with an Optima Max-XP tabletop ultracentrifuge (Beckmann Coulter) for 5 min. The supernatant was removed and the pellet was resuspended in 5.4 µl of MRB80, before further incubation for 20 min on ice. GMPCPP was then added at 1 mM final and the microtubules were polymerized at 37 °C for one additional hour. Microtubules were centrifuged at 150,000g for 5 min as described above and the supernatant was removed. Pellets were resuspended in 48 µl of MRB80 supplemented with subtilisin (1 mg ml−1) to digest the C-terminal tails of α/β-tubulin, in a subtilisin-to-tubulin final ratio of 2:1 (w/w). After a 12-min incubation at room temperature (23 ± 2 °C), Subtilisin was inhibited by addition of 10 mM PMSF (in ethanol). Next, subtilisin-treated microtubules were centrifuged at 17,000g for 10 min at room temperature to remove unpolymerized tubulin and excess subtilisin. Pellets were resuspended in MRB80 supplemented with 5% glycerol and 50 µM GMPCPP. Thereafter, resuspended subtilisin-treated microtubules were added to a reaction mixture with MRB80, 50 µM KCl, 0.025% methyl cellulose in water, 50 mM glucose, an oxygen scavenger solution (premixed; v/v/v = 1:1:1 400 µg ml−1 glucose oxidase, 200 µg ml−1 catalase and 4 mM dithiothreitol) and 50 µM GMPCPP. HiL488 tubulin was then added at a final concentration of 2 µM for 32 to 40 min at room temperature.
TIRF microscopy
Flow chambers for TIRF imaging were prepared as follows. Glass square coverslips (22 × 22 mm) were activated using O2 plasma for 1 min. Microscope slides were sonicated for 15 min in 2-propanol and rinsed with water followed by ethanol. Three bands of double-sided tape were glued to the microscope slide and coverslips were mounted on the tape. Chambers were successively filled with 20 µl of PEG-PLL-Biotin for 5 min, then 20 µl of NeutrAvidin, followed by 30 µl of k-casein and ultimately the MTD assembly reaction. Between each step, the chamber was washed once with MRB80. CenSpark-650 (2 nM in MRB80), supplemented with HiL488 tubulin (1 µM) was added 32–40 min thereafter and washed once with MRB80 before image acquisition. TIRF imaging was carried out at room temperature using an inverted microscope (Nikon, Ti-E Eclipse) equipped with a ×100 (numerical aperture: 1.49) oil-immersion objective (Nikon, Plan Apo) and a Photometrics Prime 95B camera (scientific complementary metal–oxide–semiconductor (sCMOS) grayscale chip). Illumination and image acquisition were controlled by NIS Elements Advanced Research software (Nikon). Images were captured every 30 s for 10 min with an exposure time of 100. The 638-nm, 561-nm and 488-nm laser lines were used for detecting probes, MTDs and microtubule singlets, respectively, in that order, to avoid potential crosstalk.
TIRF image analysis
Still images were processed in ImageJ as three 16-bit image channels (512 × 512 pixels). For each MTD segment considered, the background was evaluated on the full field of view for each channel by measuring the mean intensity of four distinct regions devoid of microtubules, which was then subtracted for the corresponding channel. Next, each microtubule containing a MTD segment was segmented manually on the basis of the HiL488 signal into (1) a singlet region where HiL488 signal is absent; (2) ends of singlet microtubule regions, with HiL488 signal stemming either from a MTD or from elongation of singlet microtubule; and (3) a MTD region located elsewhere, corresponding to bona fide doublet-like microtubules. We retained for further analysis singlet and MTD-like regions. The mean probe signal intensity was then determined with ImageJ (Fiji) for each region and the selectivity score for each microtubule obtained as follows:
$$\mathrm{Selectivity}\;\mathrm{score}=\frac{\mathrm{Mean}\;\mathrm{signal}\;\mathrm{intensity}\;\mathrm{at}\;\mathrm{doublet}}{\mathrm{Mean}\;\mathrm{signal}\;\mathrm{intensity}\;\mathrm{at}\;\mathrm{singlet}}$$
The selectivity score for each probe as a function of incubation time was plotted using OriginPro (version 2024b; OriginLab). Kymographs of the time-course experiments were generated using the KymographBuilder plug-in in ImageJ by manually fitting lines along the A-microtubules.
For quantitative analysis of the apparent persistence time of probes at microtubule singlets and MTDs, a binary mask of the entire microtubule was generated from the rhodamine channel image using Otsu thresholding followed by manual refinement to accurately delineate individual microtubules (mask 1). A second mask was generated by dilating mask 1 by two pixels. Next, mask 1 was subtracted from the far-red probe channel image and the larger mask 2 was applied on the same channel, identifying a circular halo of two pixels around the microtubule. The mean pixel intensity within this halo was calculated and defined as the ‘limit threshold’. Individual microtubules were then manually segmented into three-pixel-long bins along the polymer axis. Each bin was classified as belonging to a MTD region (colocalized with HiL488 signal) or a microtubule singlet region (no HiL488 signal), with microtubule ends and ambiguous singlet and MTD transition regions excluded from the analysis. For each bin, the mean pixel probe intensity was measured within the identified microtubule singlet and MTD regions. This measurement was repeated across all frames of the video. If the mean probe intensity of one bin exceeded a ‘limit threshold’ by 15%, then we considered that probe signal was present within this bin. For each bin, the residence time was then calculated as the percentage of frames in the entire video where the bin contained the probe.
Fluorescence microscopy
Initial screening of probe variants and time-lapse experiment experiments were conducted using a Zeiss Observer D1 wide-field microscope with a ×63 oil-immersion objective (NA: 1.40) and equipped with an Andor Zyla 4.2p camera. The z sections were imaged every 400 nm. A binning of 2 × 2 was applied for time-lapse experiments. In initial live imaging experiments, cells were placed in a chamber with the temperature set at 37 °C and CO2 at 5%. Stage and objective were warmed to 37 °C.
Subsequent live-cell imaging was performed using a Visitron spinning-disk CSU W1 microscope with a U Plan S-Apo ×63 oil-immersion objective (NA: 1.42), along with full temperature and CO2 control, equipped with an sCMOS grayscale camera (Orca Flash 4.0 camera, Hammamatsu Photonics), mounted on an inverted Olympus IX 83 motorized stand. The step size in z was 350 nm. Cells were maintained at 37 °C and 5% CO2 and were imaged using 405-nm, 488-nm, 561-nm and 640-nm laser lines.
Two-dimensional STED images were acquired on a Leica TCS SP8 STED 3X microscope with a ×100 (NA: 1.4) oil-immersion objective, using 488-nm, 568-nm and 656-nm excitation pulsed lasers, as well as 592-nm and 775-nm pulsed lasers for depletion.
Selectivity score quantification
To quantify interphase centriolar and cytoplasmic microtubule signals in cells, a custom image analysis pipeline was developed in ImageJ. In brief, for each three-dimensional (3D) image stack, a predetermined dark current value, specific to the sCMOS camera of the microscope, was subtracted from every pixel. Images were sum-projected and cell boundaries were identified using Cellpose52. To optimize cell detection, the projected GFP channel was transformed by taking the sixth root of each pixel intensity to reduce the influence of bright centriolar signals and enhance the weaker signal from the cell. The DNA and GFP channels were merged into a two-channel stack and Cellpose was executed using the ‘cyto2’ model with an estimated cell diameter of 50 pixels. All other Cellpose parameters were set to default values. Regions of interest (ROIs) for each cell were saved. Centrioles in the GFP channel were localized using a Laplacian of Gaussian (LoG)-based spot detection approach53. Each detected maximum was assigned to a segmented cell and, for each cell, the two brightest detected spots in the reference channel were retained, corresponding to the expected number of centrioles (note that, in S and G2, only the most intense centriole in each centrosome was considered, corresponding to the mature, mother centriole). Centrioles in the probe channel were localized using a similar LoG-based approach and associated to their GFP counterparts. To limit association error because of cell movement or channel drifts, we specified a distance of 1 µm above which the centriole signal from the two channels were not considered to be identical. For each GFP centriole localization, the brightest nearby centriole candidate marked by the probe was retained and the associated ROI was saved. ROIs of the segmented cells and centrioles detected by the probe were applied to the original 3D stack images. For each interphase cell, the mean intensity of the centriole spots detected by the probe was quantified for a defined area of 64 pixels around the local maximum on the slice with the highest signal intensity. Additionally, for each centriole, signal intensities within a concentric circle of 224 pixels around the local maximum termed the buffer area were subtracted, as this corresponds to both centriolar and cytoplasmic microtubules, overall yielding the centriole mean intensity. For each cell, a Huang dark threshold was applied on the slice from which the centriole and the buffer region were removed. The mean intensity of the remaining pixels was denoted as the microtubule mean intensity. The same principle was applied on two slices devoid of centrioles in the same cell to confirm that the mean intensity of cytoplasmic microtubules is constant within the cell. The resulting selectivity score was determined for each cell as the ratio between the centriole mean intensity and the microtubule mean intensity:
$$\mathrm{Selectivity}\;\mathrm{score}=\frac{\mathrm{Centriole}\;\mathrm{mean}\;\mathrm{intensity}\;}{\mathrm{Microtubule}\;\mathrm{mean}\;\mathrm{intensity}}$$
Fixation and immunostaining
Cells were grown on 12-mm-diameter glass coverslips, permeabilized for 15 s with 0.5% Triton X-100 in PEM (100 mM PIPES (pH 6.8), 1 mM EGTA and 2 mM MgCl2), washed twice with PEM and fixed in 0.2% glutaraldehyde (Agar Scientific, AGR1020) in PEM buffer for 15 min at room temperature. Excess glutaraldehyde was quenched for an additional 15 min using a fresh solution of NaBH4 (2 mg ml−1; Sigma-Aldrich, 213462) in PEM and washed four times with PEM. Cells were then permeabilized with 0.05% (v/v) Tween-20 for 30 min and washed in PBS and 0.05% (v/v) Tween-20, before blocking for 1 h in PBS supplemented with 0.05% (v/v) Tween-20 and 3% BSA. All antibodies were diluted in the blocking solution and incubated at room temperature for 1 h for primary antibodies and 45 min for secondary antibodies. Primary antibodies were mouse anti-GT335 (Adipogen, AG-20B-0020-C100), rabbit anti-CPAP (ProteinTech, 1517-1-AP) and mouse anti-centrin 2 (Merck, 04-1624), all diluted 1,000-fold. Secondary antibodies were goat anti-mouse Alexa Fluor 568 (Invitrogen, A11004) and goat anti-rabbit Alexa Fluor 488 (Invitrogen, A11034), both diluted 1,000-fold for confocal microscopy and 500-fold for STED microscopy. Coverslips were washed three times after each antibody incubation and further incubated with 1 μg ml−1 Hoechst 33258 (Sigma) in PBS before mounting in Fluoromount-G (Thermo Fisher Scientific, 00-4958-02). CenSpark probes were added either before fixation, following the protocol used for live-cell staining, or after fixation, with probes added to a solution of secondary antibodies in complete growth medium to obtain the desired final concentration (usually 100–500 nM). Alternatively, fixation was performed using 4% PFA in MEM and 3% PFA in MEM together with 0.02% glutaraldehyde in PEM. In these cases, the protocol for fixation, quenching and staining was identical to the one above. We eventually assessed CenSpark by performing fixation with methanol, ethanol or acetone. In this case, cells were fixed with the desired solvent for 7 min at 4 °C and washed 4 times with PEM. The protocol for staining was identical to the one above.
CLEM
RPE-1 cells were seeded on a 35-mm microgrid dish (Mattek, P35G-1.5-14-C-GRD) at a density of 30% and incubated with 6 µM RO-3306 for 17 h. Thereafter, cells were incubated for 1 h with 100 nM CenSpark and 6 µM RO-3306, washed three times and left for 75 min to complete mitosis, before imaging with a wide-field microscope as described above, using a ×63 objective to identify midbodies and a ×10 objective to register the grid of interest. The cultured cells were initially fixed for 1 h in a buffered solution of 2% paraformaldehyde and 0.5% glutaraldehyde in PBS (0.01 M, pH 7.0). Cells were then briefly washed in PBS and ROIs selected using fluorescence microscopy and then fixed again overnight in 2% paraformaldehyde and 1.0% glutaraldehyde in 0.01 M PBS pH 7.0. The next morning, cells were washed in cacodylate buffer (0.1 M, pH 7.4) and fixed in 1% osmium tetroxide with 0.8% potassium ferrocyanide in the same buffer, followed by 15 min in 0.2% tannic acid (in 0.1 M cacodylate buffer, pH 7.4). After another wash in the same buffer, cells were washed in double-distilled water and then sodium acetate at pH 5.2 for 5 min, before staining in 1% uranyl acetate in sodium acetate (pH 5.2). After a final wash in this buffer alone, cells were transferred to distilled water and dehydrated with increasing concentrations of ethanol. Once in 100% ethanol, increasing concentrations of Epon resin (Embed 812 embedding kit, EMS) were introduced until 100%. The Petri dishes were then filled with resin to a depth of resin of 2 mm, transferred to an oven at 65 °C and left for 24 h to allow the resin to cure.
Once hardened, cells of interest were identified using the alphanumeric grid pattern on the outer surface of the dish. The region was cut away and reembedded in a mold with fresh resin that was once again cured in an oven at 65 °C for 24 h. This reembedding was applied so that cells of interest and their midbodies were orientated perpendicular to the cutting plane of the diamond knife. The block was mounted in an ultramicrotome (Leica Microsystems UC6) and trimmed using glass knives to form a small trapezoid block (approximately 400 × 50 µm) around the midbody. To locate the exact ROI, repeated cutting and staining of 0.5-µm semithin sections were carried out. Finally, a series of 500 thin sections was cut (50-nm thickness) with a diamond knife (Diatome) and collected onto single-slot copper grids with a pioloform support film. These were contrasted with 2% lead citrate and 1% uranyl acetate and images were taken with a transmission electron microscope at 80 kV (Tecnai Spirit, FEI Company with an Eagle charge-coupled device (CCD) camera).
Toxicity assay
HeLa cells were seeded in Phenoplate clear-bottom 96-well plates (Revity, 6055302). After 24 h of incubation, an equal volume of complete DMEM containing Taxol, cytochalasin D, staurosporin (positive control), DMSO (negative control), doxorubicin hydrochloride (10 μM final, for total cell death) or probes was added and cells were grown in a humidified 5% CO2 incubator at 37 °C for 48 h. Afterwards, an equal volume of complete DMEM containing 2 μg ml−1 Hoechst 33342 was added and incubated in a humidified 5% CO2 incubator at 37 °C for 1 h before imaging. Cells were imaged with a GE INCell2200 automated fluorescence microscope using a Nikon ×4 (NA: 0.20), Plan Apo, CFI/60 objective, with excitation 390/18-nm and emission 432.5/480-nm filters, and images were analyzed using a custom pipeline for CellProfiler (version 4.2.7).
In vitro tubulin polymerization assay
The impact of probes on microtubule polymerization was determined using a tubulin polymerization assay monitoring changes in optical density at 340 nm (OD340) upon polymerization (Cytoskeleton, BK006P). Tubulin (3 mg ml−1; Cytoskeleton, HTS03) was incubated with compounds at different concentrations, together with 1 mM GTP in MRB80 medium. The samples were analyzed in a half-area 96-well plate (Greiner Bio-One, 675101) on an Infinite M1000 spectrofluorometer (Tecan) prewarmed at 37 °C. The excitation and emission bandwidths for all measurements were set to 20 nm and 10 nm, respectively. All samples were prepared in triplicates and fluorescence was measured over 65 min (one measurement every 20 s) until a stable signal was achieved. The lowest value of each dataset was set to 0. The obtained polymerization curves were analyzed and fitted to a Boltzmann model with a plateau followed by one-phase exponential association:
$$Y=\frac{{{\rm{A1}}}-{{\rm{A2}}}}{1+e\frac{{X}-{{\rm{X0}}}}{{{{\rm{d}}}X}}}+{{\rm{A2}}}$$
where X0 is the time at which the microtubule polymerization curve reaches its midpoint, A1 is the polymerization extent at the beginning of the reaction and A2 is the polymerization extent at the end of the reaction.
EB3–GFP dynamics in cells
RPE-1 cells were transfected with EB3–GFP54 using FuGENE 6 (LubioScience, F6-1000). Then, 1 day after transfection, cells were incubated with 100 nM CenSpark-650 for 1 h, followed by three washes with 1× PBS, supplemented with fresh DMEM and imaged. Spinning-disk confocal microscopy was performed on an inverted Nikon Eclipse Ti-E (Nikon) microscope, equipped with a perfect focus system, Nikon Plan Apo TIRF ×100 (NA: 1.49) oil objective and a spinning-disk-based confocal scanner unit (CSU-X1-A1, Yokogawa). The system was also equipped with an ASI motorized stage with the piezo plate MS-2000-XYZ (Applied Scientific Instrumentation), a Photometrics Evolve 512 electron-multiplying CCD camera (Teledyne Photometrics) and controlled by MetaMorph 7.10 (Molecular Devices). The following lasers were used: 488 nm, 150 mW (Vortran Stradus 488, Vortran Laser Technology) and 642 nm, 165 mW (Vortran Stradus 642, Vortran Laser Technology). We used ET525/50 emission filter (from ET-GFP filter set, 49002, Chroma) for imaging EB3–GFP and ET700/75 m emission filter (from ET-Cy5 filter set, 49006 Chroma) for imaging CenSpark-650. The 16-bit images were projected onto the camera chip at a magnification of 66 nm per pixel. To keep cells at 37 °C, we used a stage-top incubator (model STXG-PLAMX-SETZ21L, Tokai Hit). Microtubule plus-end tracing and kymograph plotting were performed using MetaMorph 7.10 software (Molecular Devices) as described previously55. Kymographs were further analyzed using Sigma Plot 7 software (SPSS) and data plots were prepared in Excel.
C. reinhardtii
C. reinhardtii wild-type strain CC125 mt + [137c] (Chlamydomonas Resource Center, University of Minnesota) was cultured in Tris–acetate–phosphate (TAP) medium56 under continuous white light at room temperature. Suspension cultures were maintained in Erlenmeyer flasks under constant shaking to ensure proper aeration, transferring cells to fresh TAP medium every 4 days to maintain exponential growth. For experiments, cells were transferred in Eppendorf tubes, incubated with CenSpark-555 at 500 nM and spun down. The supernatant was removed and cells resuspended in TAP medium. This was repeated twice to wash excess probe, after which cells were allowed to sit on a coverslip coated with 2 mg ml−1 poly(D-lysine) in water (Sigma-Aldrich, P6407) for 5 min, before imaging with the Visitron spinning-disk CSU W1 microscope, as above.
Human dermal fibroblasts
Human dermal fibroblasts were obtained from surgical remnants included in the VITA-certified Dermatology Biobank (CHUV_2103_12) of the Lausanne University Hospital. Informed consent was obtained from donors. Human primary dermal fibroblasts were cultured in DMEM (Thermo Fisher Scientific, 10565018) 10% FBS (Merck, A5256701) in a humidified 5% CO2 incubator at 37 °C. Cells were split every 3–4 days or at confluence. Cells were seeded in glass-bottom eight-well plates 1 day before imaging. Images were processed using ImageJ and associated plug-ins.
mES cells
CGR8 mES cells (Sigma, 07032901-1VL) were cultured on 0.1% gelatin-coated (Sigma-Aldrich, G9391) T75 flasks at 37 °C with 5% CO2 in GMEM (Sigma, G5154), supplemented with 10% ES cell-qualified FBS (Gibco, 16141-079), 1% nonessential amino acids (Gibco, 11140-050), 2 mM L-glutamine (Gibco, 25030-024), 2 mM sodium pyruvate, 100 μM 2-mercaptoethanol (Sigma, 63689-25), 1% penicillin–streptomycin (BioConcept, 4-01 F00-H), in-house-produced leukemia inhibitory factor (LIF), CHIR99021 (Merck, 361559) at 3 μM and PD184352 (Sigma, PZ0181-25MG) at 0.8 μM. Cells were passaged every 2–3 days using trypsinization (Sigma, T4049-100ML).
Then, 1 day before imaging, imaging plates were precoated with a 1:10 dilution of Biolaminin (BioLamina, LN511-0202) in DPBS containing magnesium and calcium ions (Gibco, 14040117) and cells were then plated on an µ-slide eight-well plate dish in FluoroBrite DMEM supplemented with 10% ES cell-qualified FBS, 1% nonessential amino acids, 2 mM L-glutamine, 2 mM sodium pyruvate, 100 µM 2-mercaptoethanol (Sigma-Aldrich, 63689), 1% penicillin–streptomycin, LIF, CHIR99021 at 3 µM and PD184352 at 0.8 µM. Cells were incubated for 1 h with CenSpark-650 at the desired concentration. Cells were washed with FluoroBrite DMEM before imaging.
P. multimicronucleatum
P. multimicronucleatum cells were purchased from Carolina biological supply and a clonal cell line was isolated. Paramecium cultures were fed with Chlamydomonas and cultured in cereal grass medium (1% cereal grass medium, Fisher Scientific, in Volvic water) at 27 °C. Cells were incubated with 200 nM CenSpark-650 for 1 h at room temperature in the dark in Dryll’s buffer (2 mM sodium citrate, 1 mM disodium phosphate, 1 mM monosodium phosphate and 1.5 mM calcium chloride in MilliQ water). Paramecium cells were deciliated to immobilize them by incubation in 5% ethanol in Dryll’s buffer while vortexing for 15 s. Cells were then washed in Dryll’s buffer twice to remove the remaining ethanol. Deciliated cells were identified by lack of swimming. The distance between centrioles was determined by tracing a line between their center and measuring the peak-to-peak distance. Distances between rows was determined by tracing a perpendicular line reaching the two rows of interest. For monitoring uptake into food vacuoles, P. multimicronucleatum was fed with polystyrene latex fluorescent GFP beads (Sigma-Aldrich, L5155) when the probe was added and then deciliated and imaged as above.
Primary cilia growth dynamics
RPE-1 cells were serum-starved for 24 h before imaging by replacing growth medium with DMEM/F-12 supplemented with 1% FBS. Cells were labeled with 100 nM CenSpark-650 for 1 h, washed, supplemented with 10 nM probe and then imaged by wide-field fluorescence microscopy every 10 min for 16 h. CenSpark signal length was measured manually at each time point from the base of the centriole to the ciliary tip. Length measurements were then smoothed using a three-point running average to reduce frame-to-frame variability. The time evolution of ciliary axoneme length was fitted to the analytical solution of the balance-point differential equation dL/dt = A/L − D, where A/L is the length-dependent assembly rate driven by intraflagellar transport and D is the length-independent disassembly rate, as established for Chlamydomonas flagella38,39. Steady-state cilium length was independently determined by measuring cilia length in a population of fixed serum-starved RPE-1 cells stained with CenSpark-650 (n = 11 cells).
Imaging of CAR-T cells and accompanying target cells
Live imaging of CAR-T cell immunological synapse formation was performed as described previously57. Briefly, RPMI-8226 multiple myeloma tumor cells were seeded at 50,000 cells per well in 18-well µ-slides (Ibidi,81816), allowed to adhere for 20 min and cocultured with CAR-T cells targeting B cell maturation antigen44,58, produced by retroviral transduction of healthy donor T cells, as previously described57.
T cells were stained with CenSpark-555 at 200 nM for 60 min and washed once, whereas RPMI-8226 cells were stained with CellTrace far red (Thermo Fisher Scientific, C34564) followed by three washes. Coculture was performed at a 1:1 ratio in R10 complete medium59. Imaging was conducted using a Nikon Ti2 Yokogawa CSU W1 spinning-disk confocal microscope at 37 °C, 5% CO2 and 88% humidity, with a ×40 objective and Perfect Focus System. Ten z stacks (0.5 µm apart, 5 µm total) were acquired with a frame rate of 60 s. Tumor-cell-only controls showed <2% cell death under these imaging conditions. Excitation lasers (555 nm and 647 nm) were used together with bright-field imaging. Image analysis was performed using ImageJ and custom macros.
Centriole-to-synapse distances were measured from the center of the fluorescent centrioles to the plane of contact between the CAR-T cell and the target cancer cell, obtained from the bright-field image, Cy5 residual signal and CellTrace signal. To determine the instantaneous centrosome speed, the centriole-to-synapse distances in each frame was subtracted from that in the previous frame, giving the speed in µm min−1. Hierarchical cluster analysis was performed using k-means clustering. In Supplementary Videos 7 and 8, the contour of the CAR-T cells was determined by thresholding the bright-field signal to achieve coarse-grained segmentation, before manual refinement with the aid of the cytoplasmic fluorescence signal.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.