
Human participant and clinical study details
Human volunteer recruitment and ethics approval
Volunteers were prospectively recruited under an Institutional Review Board-approved clinical protocol at the National Institutes of Health (NIH) Clinical Center (ClinicalTrials.gov Identifier NCT01568697). Participants provided written consent for inclusion in the study. Participants were reimbursed for the inconvenience of study visits. All human samples and data were collected within the NIH Clinical Center and the NIH Intramural Program. Samples and data were collected for this study between 2021 and 2024.
All enrolled participants were systemically healthy adults, screened for lack of systemic comorbidities, excluding individuals with active malignancy, history of radiation to the head and neck region, chemotherapy or radiation within the last 5 years, history of hepatitis B/C or HIV infection, autoimmune diseases and diagnosis of diabetes and/or HbA1C > 6%. Additional exclusion criteria included ongoing pregnancy or lactation, >3 hospitalizations over the last 3 years and the use of any of the following within the last 3 months: systemic antibiotics, systemic, inhaled or intranasal corticosteroids or other immunosuppressants, cytokine therapy and/or large doses of pre/probiotic supplements. Use of tobacco products over the last 1 year (including electronic cigarettes) was also an exclusion criterion.
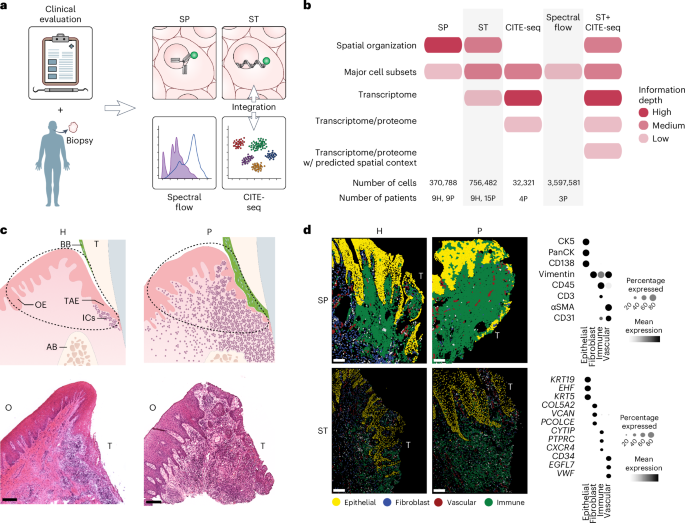
All study participants received a comprehensive oral examination, which included measurements of full mouth probing depth (PD), clinical attachment loss (CAL) and bleeding on probing (BOP) and panoramic radiography to determine periodontal status. All participants were screened for oral health, to exclude soft tissue lesions on the oral mucosa, active caries or acute periodontal/dental infections. Participants enrolled in the periodontitis group exhibited generalized inflammation (BOP > 10%), ≥4 interproximal sites with CAL ≥ 5 mm and radiographic bone loss. A total of 28 individuals met the inclusion criteria (Supplementary Table 1), consisting of 11 healthy individuals (all female; mean age 34.45 years) and 17 patients with periodontitis (7 female and 10 male; mean age 37.29 years).
Human gingiva biopsy collection
Oral biopsies of the gingiva were collected under local anesthesia from both healthy individuals and those with periodontitis. For the healthy group, biopsies were obtained from areas with no visible inflammation, no BOP and no radiographic evidence of bone loss. Periodontitis samples were collected from sites of active inflammation (BOP) and severe periodontal destruction (PD and CAL ≥ 5 mm). The exact orientation of each biopsy in relation to the tooth surface was recorded at collection and before processing. Biopsy materials that did not meet strict clinicopathologic criteria or had suboptimal orientation or tissue integrity were still included in the study but were excluded from direct quantitative comparisons between the health and periodontitis groups (Supplementary Table 1). Surgical tissue discards from periodontally involved sites obtained during periodontal surgeries were used for CITE-seq and spectral flow cytometry.
Human gingival sample preparation
Immediately following tissue acquisition, biopsy material was transferred to the laboratory, oriented and grossed into <2-mm pieces for processing according to the spatial transcriptomics (Xenium) and iterative immunostaining (IBEX) protocols. For Xenium, tissues were immersed in cryo-embedding compound (OCT) for 20 min in proper orientation at 25 °C followed by isopentane freezing. For IBEX, tissues were immersed in 1% paraformaldehyde for 72 h at 4 °C, followed by washing in 1× PBS and incubation in 30% sucrose at 4 °C overnight and were subsequently embedded in OCT.
Preparation of single-cell suspensions for CITE-seq and spectral flow cytometry was carried out as previously described52. In brief, tissue fragments were minced and digested using collagenase II (Worthington Biochemical Corporation) and DNase (Sigma) and dissociated with the gentleMACS Dissociator (Miltenyi). Cells were then passed through a 70-µm filter (Falcon, Corning), washed and counted (Cellometer Auto 2000, Nexcelom).
Spatial transcriptomics, Xenium (10x Genomics)
The 5-μm sections were placed on manufacturer-provided slides and stored at −80 °C for less than 4 weeks. Slides were then thawed for fixation, permeabilization and overnight probe hybridization followed by a post-hybridization wash, ligation, amplification, autofluorescence quenching and nuclei staining steps performed the next day. The Xenium analyzer workflow was initiated using a predesigned human multi-tissue panel (Human Multi-Tissue and Cancer) combined with a 76-plex custom probe panel (Supplementary Table 2). All steps were performed in accordance with manufacturer guidelines.
Human oral tissue iterative immunostaining, IBEX
The IBEX protocol was performed as previously described53 with minor modifications. In brief, 5-μm fixed-frozen tissue sections were placed directly onto a 25-mm round cover glass coated with 20 μl of chrome alum gelatin (Newcomer Supply). The cover glass was then adhered using low toxicity silicone elastomer material (World Precision Instruments) into a custom three-dimensional (3D) printed (Ultimaker3) chamber that allowed for consistent placement in the microscope stage insert. The chamber with the tissue sections was left to dry at room temperature inside a hood overnight. Sections were rehydrated with 1× PBS at room temperature for 5 min and blocked with 0.22 μm filtered 1× PBS containing 0.3% Triton-X-100 (VWR Life Sciences), 1% BSA (Sigma), 1% human Fc block (BD Biosciences) and 0.02% Hoechst (Biotium) at 37 °C for 1 h. Primary antibodies were incubated overnight at 4 °C, followed by secondary antibody incubation at 37 °C for 1 h, if applicable (Supplementary Table 3). The following antibodies were used: CK19 (unconjugated, mouse IgG2a, clone A53-B/A2; BioLegend 628502; 1:100 dilution), MCT (unconjugated, mouse IgG1, clone AA1; Dako M7052; 1:200 dilution), CK5 (unconjugated, guinea pig polyclonal; LS-Bio LS-C22715; 1:200 dilution), MPO (unconjugated, rabbit polyclonal; Abcam ab9535; 1:100 dilution), CD31-AF488 (mouse IgG1, clone WM59; BioLegend 303109; 1:50 dilution), Pan-CK-eF570 (mouse IgG1, clone AE1/AE3; Thermo Fisher 41-9003-80; 1:50 dilution), CD4-AF647 (mouse IgG1, clone RPA-T4; BioLegend 300523; 1:100 dilution), CD68-AF488 (mouse IgG2b, clone Y1/82A; BioLegend 333811; 1:100 dilution, not used in downstream analysis due to inconsistent quality), Ki67–eF570 (mouse IgG1, clone SolA15; Thermo Fisher 41-5698-80; 1:100 dilution), CD8α-F488 (mouse IgG1, clone RPA-T8; BioLegend 301024; 1:100 dilution), CD3-F594 (mouse IgG1, clone UCHT1; Caprico 1053134; 1:100 dilution), Thy1-AF647 (mouse IgG1, clone 5E10; BioLegend 328115; 1:100 dilution), S100A8/A9-AF488 (mouse IgG2a, clone 900028; R&D IC9337G; 1:100 dilution), α-SMA-eF570 (mouse IgG2a, clone 1A4; Thermo Fisher 41-9760-80; 1:100 dilution), CD45–iF594 (mouse IgG1, clone F10-89-4; Caprico 1016134; 1:100 dilution), CD138-AF647 (mouse IgG1, clone Syndecan-1; BioLegend 356523; 1:200 dilution), CD20-AF488 (mouse IgG2a, clone L26; Thermo Fisher 53-0202-82; 1:50 dilution), Vimentin–AF594 (mouse IgG2a, clone O91D3; BioLegend 677804; 1:600 dilution), HLA-DR-AF647 (mouse IgG2a, clone L243; BioLegend 307621; 4 °C O/N, 1:100 dilution), goat anti-mouse IgG2a AF488 (Thermo Fisher A21131; 1:500 dilution), goat anti-mouse IgG1AF555 (Thermo Fisher A21127; 1:500 dilution), goat anti-guinea pig AF647 (Thermo Fisher A21450; 1:500 dilution), goat anti-mouse IgG2b AF488 (Thermo Fisher A21141; 1:500 dilution) and donkey anti-rabbit AF647 (Jackson ImmunoResearch 711-606-152; 1:500 dilution).
Sections were equilibrated and imaged in water-soluble medium (Fluoromount G, Southern Biotech). Following image acquisition, the mounting medium was thoroughly washed off, fluorophore bleaching was performed using 1 mg ml−1 LiBH₄ (STREM Chemicals) diluted in dH2O for 30 min, and additional cycles of blocking and staining were performed. In subsequent cycles using the same isotypes as a previous indirectly labeled antibody, an additional blocking step was performed with highly concentrated (1:20 dilution) isotype controls for 1 h at 37 °C to block nonspecific antibody binding.
For visualization of HEVs, single cycle three-color immunofluorescence staining was carried out with 5-μm fixed-frozen sections placed on standard histology slides (Daigger Scientific) labeled with AF647-PNAd (clone MECA-79), iFluor594-CD3 (clone UCHT1) and AF488-CD31 (clone WM59).
IBEX image acquisition
IBEX images were acquired with a white-light laser confocal microscopy (Leica SP8) with a spectral output range between 470 nm and 670 nm equipped with an additional 405-nm laser line as well as three PMT and two HyD detectors. Images were acquired with a ×20 objective (0.75 NA). Pixel dimensions were 0.444 × 0.444 μm with a 16-bit depth. Sequential imaging between lines and bidirectional imaging were enabled. A z-stack containing the brightest slice (z-step range, 0.6–1.3 μm) was obtained. Raw outputs were then launched in LAS X software and the z slice that had the highest signal to noise ratio for all channels was selected and cropped to generate a two-dimensional image for all cycles of each sample. Huygens v.23.04 (Scientific Volume Imaging, http://svi.nl) was used for stitching of multi-tile images. Using Imaris File Converter, multichannel files from each channel were then converted to Imaris format, renamed with XTConfigureChannelSettings and registered based on Hoechst with XTRegisterSameChannel54.
IBEX image pre-processing and object-based segmentation
Registered multichannel images, were processed automatically in Fiji55. First, tissue areas that were partially unregistered or detached in any cycle were removed from all channels and all cycles via automated selection of nonoverlapping Hoechst masks in addition to manual exclusion of gross unregistered areas. Then, each individual channel was selected and processed with median filter, background subtraction, thresholding and small object elimination. For small object elimination, a mask that included positive pixels (above threshold) but also eliminated small objects for each channel (smaller than the minimum truly positive cell) was generated using the ‘Analyze Particles’ function. The mask region of interest (ROI) was superimposed to the original image and every signal outside of this ROI was removed from the channel of interest. All parameters were selected individually for each channel and kept constant across all samples and experimental batches.
For object-based segmentation, we used a combination of Fiji, Ilastik56 and Cellprofiler57. In brief, two-channel images were generated in Fiji, with the nuclei channel derived from the final cycle Hoechst channel and the cell border channel created from a composite of all membrane and cytoplasmic markers using unprocessed images (SimpleITK registration output). The two-channel stacks were then imported in Ilastik where a pixel classification project was created. Training was performed based on all samples until the probability maps for nuclei, cell borders and background were histologically accurate, and the uncertain regions were minimal. Ilastik outputs were then imported to CellProfiler to generate the segmentation masks which were used for the subsequent analysis.
IBEX cell phenotyping
Per-channel protein intensities were quantified at the single-cell level using MCQuant58 and clustering was performed using scimap59, with intensity values scaled via the ‘rescale’ function and automatic gating applied using a Gaussian mixture model. Initial clustering was carried out using the ‘phenotype_cells’ function with user-defined marker combinations, followed by subclustering within each phenotype using the k-means algorithm. When appropriate, clustering was restricted to a subset of relevant markers. For more accurate clustering of epithelial subregions, pathologist annotations were incorporated using the ‘addROI_image’ tool.
Xenium data processing and analysis
All default Xenium segmentation outputs were re-segmented using the command ‘resegment’ with an expansion distance set to 0 within the Xenium Ranger software. Xenium regions containing more than one tissue section were cropped using a custom function, such that each resulting region contained a single tissue section. In brief, a region of interest was drawn by hand in Xenium Explorer, and the region coordinates were exported as a comma-separated value file. These coordinates were used to modify the relevant raw Xenium data (cell_boundaries.csv, cells.csv, transcripts.csv and barcodes.csv) to include only coordinates within the region of interest.
Individual sections were merged into a single Seurat60 object, preserving section-specific metadata. This object was then split into individual layers representing each region. Individual regions underwent normalization followed by standard dimensionality reduction (Seurat functions: NormalizeData, FindVariableFeatures, ScaleData and RunPCA). Integration of all sections was performed using ‘IntegrateLayers’ using the RPCAIntegration method. Clustering and cluster marker genes were performed using Seurat functions (FindNeighbors, FindClusters and FindAllMarkers). Cells lacking distinct gene expression, or gene expression inconsistent with known cell types, were labeled ‘unknown’ and removed from analysis. Seurat objects were converted to AnnData format for further analysis.
Neighborhood analysis for IBEX and Xenium datasets
Spatial niche analysis was performed using the ‘spatial_expression’ tool from scimap to generate weighted neighborhood matrices for both the IBEX and Xenium datasets. Neighborhoods were defined using a k-nearest neighbors (k-NN) approach. The resulting neighborhood expression matrices were then analyzed with the ‘spatial_cluster’ tool, using k-means clustering as the dimensionality reduction method. A gradient of neighborhood sizes (number of nearest neighbors) and a range of cluster numbers (k) were applied iteratively to optimize niche resolution. For final clustering, 30 nearest neighbors were used for IBEX and 50 for Xenium, reflecting differences in tissue architecture and spatial resolution. When necessary, niche clusters were manually merged based on shared cell-type composition and histologic context.
Spatial proximity analysis of immune niches to TAE
To assess the spatial proximity of immune cell niches to the TAE niche, a custom Python script was used, utilizing scikit-learn for pairwise Euclidean distance calculations between cells based on their X and Y centroids. The analysis was restricted to sections with ≥200 TAE cells to ensure robust results. Pairwise distances were calculated between TAE and immune cells. A maximum distance threshold of 3,000-pixel units was applied to account for variation in section size and to exclude distant areas that appeared only in larger sections. For each image and niche, the analysis quantified (1) the number of immune cells within the 3,000-unit threshold and (2) the average distance between TAE and immune niche cells.
CITE-seq
Single-cell preparations of between 5 × 104 and 5 × 105 single cells from periodontitis samples were stained individually with TotalSeq-A Human Universal Cocktail, v.1.0 (BioLegend) as per the manufacturer’s protocol (Supplementary Table 4). The 1.7 × 104 stained cells were then loaded with the reverse transcription mix on the Chromium chip G and partitioned into single cells in gel beads-in-emulsion (GEMs) using the 10x Genomics Chromium Next GEM Single Cell 3’ kit v3.1, as per the manufacturer’s instructions (10x Genomics). Complementary DNA amplification and ADT libraries were prepared as per instructions from BioLegend, and gene expression libraries were made as per the 10x Genomics protocol. All amplification steps were performed in an Applied Biosystems Instruments Veriti 96-well thermal cycler. Quality and quantity of the libraries were assessed using TapeStation (Agilent) and a Qubit fluorometer (Thermo Fisher). Libraries were pooled at a concentration of 2 nM and sequenced on an Illumina platform (NextSeq 2000, Illumina) using the read lengths as read 1, 28 bp; index 1, 10 bp; index 2, 10 bp; and read 2, 90 bp.
Processing of CITE-seq data was performed in R using DecontX to correct for ambient RNA, DSB to normalize and denoise ADT counts and DoubletFinder to remove putative doublets. Seurat was used for data normalization, determination of variable features, data scaling and principal component analysis (PCA). RPCA integration was performed on RNA and ADT assays and a WNN graph was created using the Seurat FindMultiModalNeighbors. General cell types were identified using FindClusters and FindAllMarkers functions before subsetting the integrated data by cell type. Variable feature identification, scaling, PCA, RPCA integration and WNN graph creation were then performed within each cell type to identify cell type subsets.
Spectral flow cytometry
Freshly isolated oral tissue samples yielding between 7 × 105 and 2 × 106 single cells were processed for flow cytometric analysis using a 36-color panel (Supplementary Table 5). Samples were first stained with Live/Dead reagent for 15 min at 25 °C, washed and incubated with Fc block for 10 min at 25 °C. Cells were next incubated with a pool of six panel antibodies for 10 min at 25 °C: CCR5, CCR6, CCR7, CXCR3, CXCR5 and TCRgd. A pool of all remaining panel antibodies was then added and samples incubated for additional 30 min at 25 °C. Following this, cells were washed and fixed with 1% paraformaldehyde for 10 min at 25 °C. Acquisition was conducted using a Cytek Aurora with spectral unmixing and autofluorescence extraction using SpectroFlo software. For unmixing, single color reference controls were used of each antibody staining beads (UltraComp eBeads Plus, Thermo Fisher) and viability dye staining in a mixture of live and heat-killed peripheral blood mononuclear cells. For autofluorescence extraction, controls of unstained matching oral tissue sample were used. Data analysis was performed in FlowJo.
CITE-seq and Xenium integration for spatial imputation of cluster identities
Computational integration between CITE-seq and Xenium data was performed to spatially impute fine-grained cluster identities from CITE-seq into Xenium samples. In brief, the CITE-seq Seurat objects were converted to AnnData format. Xenium and CITE-seq AnnData objects were subset into broad cellular compartments (epithelial, stromal and immune) and each compartment was processed independently. Each dataset was concatenated, scaled and underwent PCA, followed by batch correction using the ‘scanpy.external.pp.harmony_integrate’ function. Cell-type labels from CITE-seq were then transferred to Xenium using k-NN classification based on the harmonized PCA embeddings. Finally, the integrated epithelial, stromal and immune datasets were merged into a single AnnData object for downstream analysis.
Barrier epithelial cell cross-organ integration and reanalysis
Gingival epithelial cells from the human oral single-cell atlas13 were integrated with epithelial cells from the Xenium dataset for label transfer from the Xenium to the single-cell dataset. Label-transferred epithelial cells from healthy gingiva were then concatenated with epithelial cells from the Tabula Sapiens reference atlas17 for cross-organ comparison. The merged dataset was then normalized, log-transformed and reduced to the top 2,000 highly variable genes. Data rescaling and PCA were performed, followed by batch correction using ‘scanpy.external.pp.harmony_integrate’. For cross-organ comparison, the integrated dataset was further filtered to include only epithelial cells from mucosal barrier tissues (oral mucosa, gastrointestinal tract and lung), while excluding nonbarrier epithelial subsets, such as secretory or neuroendocrine cells. The top 30 defining genes for each organ were submitted to Enrichr51 to identify the most enriched Gene Ontologies specific to each mucosal barrier tissue.
Bacterial FISH and immunofluorescence labeling on human gingival tissues
FISH was performed using eubacterial FISH probe (GCTGCCTCCCGTAGGAGT) conjugated to rhodamine-red X fluorophore (Integrated DNA Technologies) on 5-μm fixed-frozen sections. Hybridization was carried out at 46 °C O/N in hybridization buffer (0.09 M NaCl, 0.02 M Tris, pH 7.5, 0.01% SDS and 20% formamide) and 2 µM EUB probe. Slides were washed in wash buffer 1 (0.09 M NaCl, 0.02 M Tris, pH 7.5, 0.01% SDS and 20% formamide) for 15 min at 48 °C followed by three washes in wash buffer 2 (0.09 M NaCl, 0.02 M Tris, pH 7.5 and 0.01% SDS) for 15 min at 48 °C. Slides were dehydrated in an ethanol series before proceeding to immunofluorescence labeling. For CK19 immunofluorescence labeling, the sections were permeabilized for 10 min with PBS containing 0.1 % Tween 20. Followed by three PBS washes. To block nonspecific binding of antibody, sections were incubated with 1% BSA, 22.52 mg ml−1 glycine in PBST for 30 min. Sections were then incubated in the diluted (1:100) primary antibody (clone A53-B/A2) for 1 h at 25 °C. The slides were washed three times for 5 min each with PBS. The sections were further incubated with the anti-mouse IgG2a conjugated to AF488 (1:100) for 1 h in the dark. The slides were washed three times for 5 min each with PBS. The sections were then counter stained with 3.0 μg ml−1 4,6-diamidino-2-phenylindole (DAPI) for 5 min and rinsed with PBS. The slides were mounted in Prolong Gold antifade medium (Thermo Fisher) and allowed to cure for at least 24 h before imaging.
Images were acquired using the LSM 980 confocal microscope (Carl Zeiss). The spectral images were acquired with the Plan-Apochromat ×20/0.8 NA for a full tile image of the entire tissue field of view and Plan-Apochromat ×63/1.4 NA for hi-res of regions of interest. The 24-channel spectral images were acquired by simultaneous excitation with 488 nm and 561 nm lasers and signals collected from 490–695 nm. The 405 nm laser was used independently to image DAPI in a separate image. The 3D z-stack images were acquired with 7–9 z-planes up to a thickness of 7 μm. Spectral unmixing was performed in Zenv v.3.6 and image brightness and contrast were adjusted for each unmixed channel separately in Fiji ImageJ v.1.54F.
Statistics and reproducibility
No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to or exceed those reported in previous publications13,17,43. Unless otherwise indicated, statistical comparisons were performed using nonparametric tests, as the datasets did not meet the assumptions of normality required for parametric testing. Normality and equal variance were not formally tested.
All selected photomicrographs are representative of spatial patterns reproducibly observed across all appropriately oriented samples and experimental batches (spatial proteomics, n = 8 (oral health) and n = 8 (periodontitis) participants across three independent experiments; spatial transcriptomics, n = 8 (oral health) and n = 11 (periodontitis) participants across two independent experiments).
Data collection and analysis were not performed blind to the conditions of the experiments, but analysis was performed using automated and unsupervised methods. No subjective input influenced the analysis outputs. No randomization occurred during sample collection or analysis.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.