
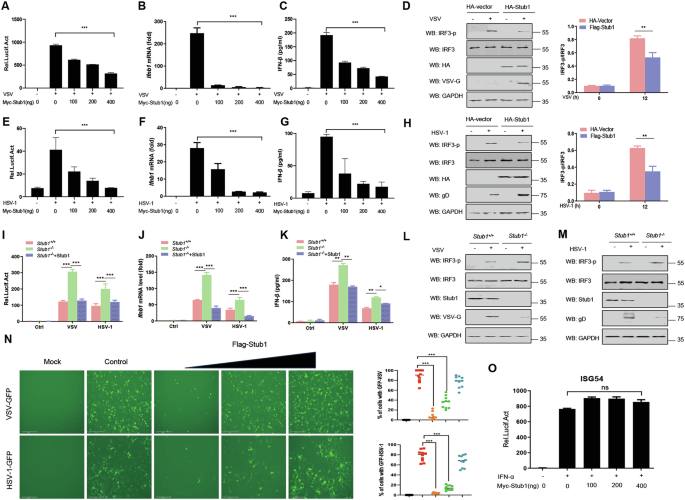
Stub1 negatively regulates type I IFN signaling
To assess the effects of Stub1 on RIG-I- and cGAS-STING-mediated type I IFN signaling pathways, HeLa cells were transfected with the indicated luciferase reporters and then infected with the RNA virus vesicular stomatitis virus (VSV) or the DNA virus herpes simplex virus type I (HSV-1). Our results indicate that ectopic expression of Stub1 significantly inhibits VSV infection-mediated IFN-β production (Fig. 1A–C) as well as that of HSV-1 (Fig. 1E–G). In addition, Stub1 inhibited the activation of IFN-β induced by intracellular (IC) poly(I:C) as well as poly (dA: dT) treatment (Fig. S1A–D). Furthermore, we found that the overexpression of Stub1 inhibited the activities of promoters, including those of IFN-α, IFN-stimulated response element (ISRE), and the IFN-stimulated factor ISG54, induced by VSV and HSV-1 infection in a dose-dependent manner (Fig. S1E–J). Consistently, Stub1 inhibited the phosphorylation of IRF3 during VSV and HSV-1 infection (Fig. 1D, H).

HEK293T cells were transfected with an IFN-β luciferase reporter and a Renilla-TK reporter, together with increasing amounts (0, 100, 200, and 400 ng) of a plasmid expressing Myc-Stub1 for 12 h, and then the cells were infected with VSV (0.1 MOI) (A) or HSV-1 (10 MOI) (E) for another 12 h. Luciferase activities were then analyzed. HEK293T cells were transfected with increasing amounts (0, 100, 200, or 400 ng) of a plasmid expressing Myc-Stub1 for 12 h, after which the cells were infected with VSV (0.1 MOI) (B) or HSV-1 (10 MOI) (F) for another 12 h, and the mRNA levels of Ifnb1 were subsequently analyzed via qPCR. C, G ELISA of IFN-β in the supernatant shown in (B, F). HeLa cells were transfected with a plasmid expressing Myc-Stub1 for 12 h, after which the cells were left uninfected or infected with VSV (0.1 MOI) (D) or HSV-1 (10 MOI) (H). Then, IRF3, phosphorylated IRF3, the VSV protein, and GAPDH were analyzed by immunoblotting. The band intensity of the IRF3-p/IRF3 immunoblotting result was quantified via ImageJ. I HEK293T-stub1+/+ and HEK293T-stub1−/− cells were transfected with an IFN-β Luc reporter, a Renilla-TK reporter, and an empty vector or a plasmid expressing Flag-Stub1. The Luc activity of the IFN-β promoter-reporter was detected. J HEK293T-stub1+/+ and HEK293T-stub1−/− cells were transfected with an empty vector or a plasmid expressing Flag-Stub1. The mRNA levels of Ifnb1 in the cells were subsequently analyzed via qPCR. The results are presented relative to those of untreated cells transfected with an empty vector. K ELISA of IFN-β in the supernatant shown in (J). HEK293T-stub1+/+ and HEK293T-stub1−/− cells were infected with VSV (0.1 MOI) (L) or HSV-1 (10 MOI) (M) for 12 h and then subjected to immunoblot analysis with antibodies against IRF3, phosphorylated IRF3, VSV protein, HSV-1 protein, and Stub1. N An IFN sensitivity assay was performed to test the inhibitory effects of Stub1 on the replication of VSV-GFP (0.1 MOI) or HSV-1-GFP (10 MOI). VSV-GFP and HSV-1-GFP replication was analyzed via fluorescence microscopy, and quantitative analysis was performed by measuring the proportion of GFP-positive cells. O Luciferase activity of the ISG56-Luc reporter in HEK293T cells transfected with a Renilla-TK reporter together with different amounts (0, 100, 200, and 400 ng) of a plasmid expressing Myc-Stub1 and then stimulated with IFN-α (500 IU/mL) for 12 h. Ns not significant (P > 0.05), *0.01 < P < 0.05, **P < 0.01, and ***P < 0.001 (two-tailed Student’s t test). The data are representative of three independent experiments with three biological replicates (the mean ± standard deviation of triplicate assays or are representative of three independent experiments with similar results) (A–O).
To further validate these observations, a stub1 gene knockout (KO) HEK293T cell line (293T-stub1−/−) was generated via CRISPR-Cas9 to examine the function of Stub1 in vitro. We found that VSV or HSV-1 infection increased IFN-β production in Stub1-deficient cells (Fig. 1I–K). Moreover, the phosphorylation levels of IRF3 in HEK293T-stub1−/− cells were greater than those in HEK293T-stub1+/+ cells infected with VSV or HSV-1, although the total protein levels of IRF3 were not affected (Fig. 1L, M). To further demonstrate the role of Stub1 in type I IFN signaling, an IFN sensitivity assay was performed. As shown in Fig. 1N, Stub1 inhibited the replication of VSV-GFP and HSV-1-GFP by suppressing the production of IFN-I. To test whether Stub1 has a potential effect on the IFN signaling pathway, we assessed the effect of Stub1 on ISG56 promoter activity in IFN-treated cells. The results demonstrated that Stub1 did not affect IFN-induced ISG56 promoter activity (Fig. 1O).
To study the effects of Stub1 on host antiviral responses, especially type I IFN production in vivo, Stub1fl/fl mice and Stub1fl/fl Lyz2-Cre mice were generated and validated by sequencing and Western blot analysis (Fig. S2A–E). As demonstrated in Fig. 2A, B, primary bone marrow-derived macrophages from Stub1fl/fl mice and Stub1fl/fl Lyz2-Cre mice were treated with poly(I:C), poly (dA: dT), or infected with EMCV, VSV, PRV, or HSV-1, respectively. A comparison of primary bone marrow-derived macrophages (BMDMs) from Stub1fl/fl mice with those from Stub1fl/fl Lyz2-Cre mice revealed that the former presented reduced mRNA expression of Ifnb1 and Isg56. In accordance with these findings, the protein levels of IFN-β in the culture medium of primary bone marrow-derived macrophages from Stub1fl/fl Lyz2-Cre mice were also found to be considerably elevated.

A, B Primary bone marrow-derived macrophages isolated from Stub1fl/fl mice and Stub1fl/fl Lyz2-Cre mice were transfected with poly(I:C) (1 μg/mL) or poly (dA: dT) (1 μg/mL) or infected with VSV (1 MOI), EMCV (1 MOI), HSV-1 (10 MOI), or PRV (1 MOI) for 12 h. The mRNA levels of Ifnb1 (A) and Isg56 (B) were analyzed via qPCR. C ELISA of IFN-β in the supernatant shown in (A, B). qPCR analysis of the mRNA levels of Ifnb1 (D), Isg56 (E), and Mx1 (F), the genomic copy numbers (G) and the viral titers measured by TCID50 assay of VSV (H) in peritoneal macrophages isolated from Stub1fl/fl mice and Stub1fl/fl Lyz2-Cre mice infected with VSV (MOI of 1) for 0, 4, 8, or 12 h. qPCR analysis of the mRNA levels of Ifnb1 (I), Isg56 (J), and Mx1 (K) and the genomic copy numbers (L) and the viral titers measured by a TCID50 assay of HSV-1 (M) in peritoneal macrophages isolated from Stub1fl/fl mice and Stub1fl/fl Lyz2-Cre mice infected with HSV-1 (MOI of 10) for 0, 4, 8, or 12 h. qPCR analysis of the mRNA levels of Isg56 (N) and Mx1 (O) in BMDMs isolated from Stub1fl/fl mice and Stub1fl/fl Lyz2-Cre mice stimulated with IFN-α (500, 1000, or 2000 IU/mL) for 12 h. qPCR analysis of the mRNA levels of Isg56 (P) and Mx1 (Q) in peritoneal macrophages isolated from Stub1fl/fl mice and Stub1fl/fl Lyz2-Cre mice stimulated with IFN-α (500, 1000, 2000 IU/mL) for 12 h. Data are representative of three independent experiments with three biological replicates (mean ± SD). NS not significant (P > 0.05), **P < 0.01, ***P < 0.001.
To determine whether the mRNA expression of Ifnb1 could be affected by Stub1, peritoneal macrophages isolated from Stub1fl/fl mice and Stub1fl/fl Lyz2-Cre mice were infected with VSV or HSV-1 for 0, 4, 8, or 12 h. Compared with peritoneal macrophages derived from Stub1fl/fl mice, peritoneal macrophages from Stub1fl/fl Lyz2-Cre mice infected with the two viruses presented increased mRNA expression of Ifnb1, Isg56, and Mx1 at various time points (Fig. 2D–F, H–J). Furthermore, the viral genomic copy number and titer in peritoneal macrophages from Stub1fl/fl Lyz2-Cre mice were significantly lower than those in peritoneal macrophages from Stub1fl/fl mice (Fig. 2G, H, K–M). Consistent with the results shown in Fig. 1O, the results of the qPCR analysis revealed that Stub1 knockout did not affect the mRNA levels of IFN-induced Isg56 or Mx1 in either BMDMs or peritoneal macrophages isolated from Stub1fl/fl mice or Stub1fl/fl Lyz2-Cre mice after IFN-α treatment (Fig. 2N–Q). These collective results indicate that the knockout of Stub1 resulted in an increase in the production of type I IFNs and ISGs, thereby inhibiting viral replication.
To directly test whether the enhanced antiviral state resulting from Stub1 deficiency is mediated by IFN-I signaling, we isolated BMDMs from Stub1fl/fl mice or Stub1fl/fl Lyz2-Cre mice and performed IFNAR antibody blockade experiments. Treatment with anti-IFNAR antibodies abrogated the differences in IFN-β induction, ISG expression, and viral replication between Stub1-KO and wild-type macrophages (Fig. S3A–C), confirming the essential role of IFN-I signaling in this phenotype. Furthermore, we assessed the specificity of Stub1’s action. Stub1 deficiency enhanced Ifnb1 mRNA production induced by both the viral mimic poly(I:C) and the bacterial LPS, but did not affect the expression of the proinflammatory cytokine Tnf-α (Fig. S3D, E), indicating that Stub1 selectively inhibits the TRIF-TBK1-IRF3 axis without affecting the MyD88-NF-κB pathway, consistent with its TBK1-centric mechanism.
Stub1 suppresses type I IFN signaling by targeting TBK1
To clarify the underlying mechanisms by which Stub1 negatively regulates IFN-I production, we assessed the effect of Stub1 on IFN-β reporter activation induced by key molecules in the RIG-I and cGAS-STING signaling pathways in HEK293T cells. As shown in supplementary Fig. S3A–G, ectopically expressed Stub1 significantly inhibited the IFN-β reporter activation induced by RIG-I, MDA5, MAVS, TBK1, and cGAS+STING in a dose-dependent manner but not by IKKƐ or IRF3-5D (a constitutively active variant of IRF3) (Fig. S4A–G). In accordance with these results, qPCR analysis revealed that the ectopic expression of Stub1 inhibited the IFN-β reporter activation of Ifnβ1 induced by RIG-I, MDA5, MAVS, TBK1, and cGAS+STING but not by IKKε or IRF3-5D (Fig. 3A). Subsequently, HEK293T-stub1−/− and HEK293T-stub1+/+ cells were transfected with plasmids expressing RIG-I, MDA5, cGAS+STING, MAVS, TBK1, or IKKε and IRF3-5D. We found that IFN-β reporter activation was significantly increased by RIG-I, MDA5, MAVS, TBK1, and cGAS+STING in HEK293T-stub1−/− cells compared with that in HEK293T-stub1+/+ cells but not by IKKε or IRF3-5D (Fig. 3B). Taken together, these results indicate that Stub1 may negatively regulate IFN-I production by targeting TBK1.

A HEK293T cells were transfected with an IFN-β luciferase reporter and a Renilla-TK reporter and a plasmid expressing RIG-I, MDA-5, MAVS, TBK1, IKKε, IRF3-5D, or cGAS+STING, along with an empty vector or a plasmid expressing Flag-Stub1. The Luc activity of the IFN-β promoter-reporter was detected. B HEK293T-stub1+/+ and HEK293T-stub1−/− cells were transfected with an IFN-β luciferase reporter, a Renilla-TK reporter, and a plasmid expressing RIG-I, MDA-5, MAVS, TBK1, IKKε, IRF3-5D, or cGAS+STING. The luciferase activity of the IFN-β promoter reporter was detected. C HEK293T cells were transfected with plasmids expressing Myc-Stub1 alone or together with plasmids expressing Flag-RIG-I, MDA5, MAVS, TBK1, IKKε, IRF3, cGAS, or STING for 24 h. The cell lysates were harvested and used for Co-IP. Input and IP complexes were analyzed by Western blotting using antibodies against Flag and Myc. D HEK293T cells were transfected with plasmids expressing HA-Stub1 alone or together with a plasmid expressing Flag-TBK1. At 24 hpt, the cell lysates were harvested and used for reverse Co-IP. Input and IP complexes were analyzed by Western blotting using antibodies against Flag and HA. E His-TBK1 was incubated with GST or GST-Stub1 for 30 min, and the protein-protein interactions were then confirmed via a GST pull-down assay. F Bimolecular fluorescence complementation (BIFC) analysis of TBK1 and Stub1. HA-Stub1-VN, Stub1 fused with the N-terminus of Venus; Flag-TBK1-VC, TBK1 fused with the C-terminus of Venus. G, H Co-IP and immunoblot analysis of the interaction of endogenous Stub1 and TBK1 in THP-1 cells infected with VSV (0.1 MOI) (F) or HSV-1 (10 MOI) (G) for 12 h. Co-IP was subsequently performed with an anti-stub1 antibody. IgG was used as a negative control. I THP-1 cells were mock-infected or infected with VSV (0.1 MOI) or HSV-1 (10 MOI) for 12 h. The subcellular localization of endogenous Stub1 and TBK1 was analyzed via fluorescence microscopy. Ns not significant (P > 0.05), *0.01 < P < 0.05, **P < 0.01, ***P < 0.001 (two-tailed Student’s t test). The data represent three independent experiments with three biological replicates (means ± SDs) or three independent experiments with similar results (A–I).
To further identify the targets of Stub1, we tested the interactions between stub1 and RIG-I, MDA5, MAVS, TBK1, IKKε, IRF3, cGAS, and STING. Coimmunoprecipitation (co-IP) results revealed that Stub1 interacted with RIG-I, MAVS, and TBK1 but not with the other proteins (Fig. 3C). Because RIG-I and MAVS have been investigated by other researchers [39, 40], we focused on TBK1 in this study. As shown in Fig. 3D, Stub1 coimmunoprecipitated with TBK1 when the indicated proteins were co-expressed in HEK293T cells. To determine whether Stub1 physically interacted with TBK1 in vitro, we performed pull-down assays and found that His-TBK1 could be pulled down by GST-Stub1 but not by GST (Fig. 3E). We further demonstrated the interaction between Stub1 and TBK1 via bimolecular fluorescence complementation analysis (BIFA) (Fig. 3F). In addition, we also found that endogenous Stub1 interacted with endogenous TBK1 in HEK293T cells following both VSV infection and HSV-1 infection (Fig. 3G, H). Interestingly, when the lysosomal inhibitor CQ is added under conditions conducive to viral infection, the interaction between Stub1 and TBK1 is enhanced. These findings demonstrate that Stub1 plays a role in stabilizing TBK1 expression under such conditions (Fig. 3G, H). Immunofluorescence staining also revealed that Stub1 colocalized with TBK1 in the cytoplasm (Fig. 3I). To further substantiate that Stub1 limits IFN-I production primarily by targeting TBK1, we employed three distinct viral infection models alongside knockdown of key signaling molecules. Notably, while knockdown of RIG-I or MAVS did not abolish the inhibitory effect of Stub1 on IFN-β production induced by VSV, EMCV, or HSV-1, knockdown of TBK1 almost completely abolished this inhibition (Fig. S5A–G). These results reinforce the conclusion that Stub1 suppresses type I interferon production predominantly via TBK1.
Moreover, Co-IP revealed that Stub1 interacted with the ULD (ubiquitin-like domain) of TBK1 through its U-box domain (Fig. S6A–D). Collectively, these results suggest that Stub1 suppresses type I IFN signaling by targeting TBK1.
Stub1 promotes the autophagic degradation of TBK1
Stub1 can promote the degradation of RIG-I [15] and MAVS [40]. We first assessed the effects of Stub1 on the degradation of RIG-I, TBK1, and MAVS in HEK293T cells. We found that increasing amounts of Stub1 significantly decreased the protein levels of RIG-I and TBK1 but not those of MAVS (Fig. 4A). Furthermore, we did not observe significant Stub1-induced MAVS degradation in HEK293T and HeLa cell systems (Fig. S7A–C). This suggests that Stub1’s regulatory function may be highly context-dependent, potentially varying across cell types, species, or specific cellular states. However, the mRNA level of TBK1 remained unchanged (Fig. 4C), suggesting that Stub1 promotes TBK1 protein degradation (Fig. 4B). The CHX-chase assay results revealed that, compared with the control, Stub1 overexpression reduced the TBK1 half-life in HEK293T cells (Fig. 4D). Furthermore, the knockout of Stub1 resulted in TBK1 stability during VSV infection (Fig. 4E). The CHX-chase assay results revealed that the degradation rate of TBK1 in 293T-stub1−/− cells was lower than that in WT cells (Fig. 4F, G).

A HEK293T cells were transfected with increasing amounts (0, 0.5, 1, and 2 µg) of a plasmid expressing HA-Stub1, and the protein was harvested for immunoblot analysis. B, C HEK293T cells were transfected with Flag-TBK1, together with increasing amounts (wedge) of HA-Stub1 expression vector, and the protein was harvested for immunoblot analysis. Flag-TBK1 expression was normalized to that of GAPDH (below) (B). C Right, RT-PCR analysis of TBK1 mRNA; GAPDH mRNA served as a loading control. D Immunoblot analysis of protein extracts from HEK293T cells transfected with Flag-TBK1 or HA-Stub1 and treated with cycloheximide (CHX) (100 μg/mL) for the indicated durations (0, 4, 8, or 12 h). Flag-TBK1 expression was normalized to that of GAPDH (below). E Protein lysates of HEK293T-stub1+/+ and HEK293T-stub1−/− cells infected with VSV (MOI = 0.1) at the indicated time points were immunoblotted with the indicated antibodies. F, G Immunoblot analysis of protein extracts from HEK293T-stub1+/+ and HEK293T-stub1−/− cells treated with cycloheximide (CHX) (100 μg/mL) for the indicated durations. H Immunoblot analysis of protein lysates of HEK293T cells transfected with vectors expressing Myc-Stub1-WT, Myc-Stub-K31A, Myc-Stub-H261A, or the Myc-Stub1-DM mutant, together with plasmids encoding Flag-TBK1. I Analysis of the Luc activity of the IFN-β promoter reporter in HEK293T cells transfected with vectors expressing Myc-Stub1-WT, Myc-Stub-K31A, Myc-Stub-H261A, or the Myc-Stub1-DM mutant together with plasmids encoding Flag-TBK1. J HEK293T cells were transfected with a plasmid encoding Flag-TBK1 together with plasmids encoding EV or Myc-Stub1, followed by treatment with MG132 (10 μM), 3MA (10 mM), CQ (50 μM), or bafilomycin A1 (Baf A1) (0.2 μM) for 6 h. The cell lysates were then analyzed by immunoblotting. Flag-TBK1 expression was normalized to that of GAPDH (below). K Protein lysates of WT, BECLN1 KO, ATG5 KO, and ATG7 KO 293T cells transfected with plasmids encoding EV or HA-Stub1. The data represent three independent experiments with three biological replicates (means ± SDs) or three independent experiments with similar results (A–K). Statistical significance was determined by two-tailed Student’s t test or one-way ANOVA followed by the Bonferroni post hoc correction, as appropriate. *P < 0.05, **P < 0.01, ***P < 0.001.
To investigate whether the enzyme activity of Stub1 is required to promote TBK1 degradation, we generated four plasmids expressing catalytically inactive Stub1 mutants, namely, Stub1-wt, Stub1-K31A, Stub1-H261A, and Stub1-DM (double mutants K31A and H261A). Stub1-K31A has a point mutation in the TPR chaperone-binding domain, which prevents it from binding to chaperones; consequently, its ability to bind substrates is greatly reduced. Stub1-H261A has a point mutation in the U-box domain of Stub1 and, therefore, is deficient in E3 ubiquitin ligase activity; thus, its ability to ubiquitinate the substrate for degradation is diminished [43]. We observed that the TBK1 protein was no longer degraded when only TBK1 and Stub1-DM were co-expressed in the cells (Fig. 4H). These results suggest that Stub1 may inhibit IFN-I production through its enzymatic activity. The results of the dual-luciferase reporter gene assay demonstrated that Stub1 inhibits TBK1-mediated IFN-β promoter activity through its E3 ubiquitin ligase enzymatic activity and chaperone binding ability (Fig. 4I). Our research revealed that when Stub1 undergoes a single-site mutation, it cannot fully restore TBK1 expression. However, when the molecular chaperone-binding site of Stub1 is mutated, and the autophagy-related protein Beclin1 is simultaneously knocked out, TBK1 expression is fully restored (Fig. S7D). Therefore, we hypothesize that Stub1 may promote the ubiquitination of TBK1 through its E3 ubiquitin ligase activity, leading to its degradation via the autophagy pathway. Notably, when the E3 ubiquitin ligase activity site of Stub1 is mutated, and the molecular chaperone protein HSC70 is simultaneously knocked out, TBK1 expression is fully restored (Fig. S7E). Therefore, we hypothesize that other E3 ubiquitin ligases may promote TBK1 K27-linked ubiquitination, followed by interaction with HSC70 to achieve degradation. To establish a direct correlation between phenotype and the TBK1-CMA axis, BMDMs were isolated from Stub1fl/fl mice and Stub1fl/fl Lyz2-Cre mice. The Stub1-WT gene and its double-site inactivating mutant Stub1-DM were reintroduced into the cells via lentivirus, and TBK1 expression was detected via Western blot analysis. The results demonstrated that TBK1 expression recovered to stable levels following Stub1 deletion. Reoverexpression of wild-type Stub1 led to a significant reduction in TBK1 expression, whereas overexpression of the Stub1-DM plasmid, which carries an enzyme active site mutation, resulted in the restoration of TBK1 expression to stable levels (Fig. S7F).
Protein degradation primarily occurs via two pathways: the proteasome-dependent pathway and the lysosome-dependent pathway [44]. To further investigate Stub1-mediated TBK1 degradation, Stub1 and TBK1 were co-expressed in HEK293T cells, which were then treated with a proteasome inhibitor (MG132), a lysosomal inhibitor (chloroquine (CQ)), a phosphatidylinositol 3-kinase inhibitor (3-methyladenine (3-MA)), or an autophagosome-lysosome fusion inhibitor, bafilomycin A1 (Baf-A1). As shown in Fig. 4J, only CQ treatment suppressed TBK1 degradation. Consistently, Stub1 was still able to trigger TBK1 degradation after Stub1 was overexpressed in ATG5-, ATG7-, and Beclin1-KO cells (Fig. 4K). We also incorporated inhibitors such as CQ and MG132 into time-dependent CHX-mediated degradation assays. The results revealed that TBK1 protein expression recovery was observed exclusively in chloroquine-treated cells (Fig. S7G). Taken together, these results suggest that Stub1 specifically degrades TBK1 via the lysosomal pathway during viral infection.
Stub1 promotes the autophagic degradation of TBK1 by enhancing K27 polyubiquitination at the K344 site of TBK1
The E3 ubiquitin ligase activity of Stub1 has been implicated in TBK1 degradation. To determine whether Stub1 directly ubiquitinates TBK1, we examined the effect of Stub1 overexpression on TBK1 polyubiquitination and observed a significant increase. Further analysis revealed that Stub1 specifically promotes K27-linked polyubiquitination of TBK1, with no apparent effect on other types of ubiquitin linkages (Fig. S8A). We then conducted in vitro ubiquitination experiments to elucidate the role of Stub1 in the ubiquitination process of TBK1. The smeared bands detected by ubiquitin antibodies following Western blotting indicated that polyubiquitin chains of different molecular weights had formed (Fig. S8B). In contrast, no smeared bands were detected in the reaction containing E1, E2, or His-TBK1 but lacking GST-Stub1 (Fig. S8B), indicating that Stub1 is a specific ubiquitin ligase for TBK1. Stub1 carrying mutations in the ubiquitin ligase active site, such as Stub1-H261R or Stub1-DM, lost ubiquitin ligase activity, as no diffuse bands of polyubiquitin were detected in these reactions (Fig. S8C), supporting the importance of Stub1 enzyme activity for TBK1 ubiquitination. In addition, we found that TBK1 underwent K27-linked polyubiquitination approximately 4 h after viral infection, which was markedly enhanced by the overexpression of Stub1 (Fig. 5A). We further explored the role of the enzymatic activity of Stub1 in promoting the K27-linked polyubiquitination of TBK1 and revealed that the overexpression of Stub1 and Stub1-K31A, but not Stub1-H261A, promoted the K27-linked polyubiquitination of TBK1 (Fig. 5B, C). Consistently, the depletion of Stub1 resulted in decreased K27-linked polyubiquitination of TBK1 during VSV infection (Fig. 5D). To further confirm that endogenous TBK1 undergoes covalent modification via K27-linked polyubiquitin chains, we immunoprecipitated endogenous TBK1 from Stub1fl/fl and Stub1fl/fl Lyz2-Cre BMDMs. Western blot analysis of immunoprecipitates with a K27-linkage-specific antibody revealed robust infection-induced K27-linked polyubiquitination of TBK1 in control cells, which was significantly attenuated in Stub1-deficient macrophages (Fig. 5E). In contrast, Stub1 deficiency had no significant effect on K48-linked ubiquitination. Together, these data provide direct endogenous evidence that Stub1 specifically mediates K27-linked polyubiquitination of TBK1.

A Protein lysates of HEK293T cells transfected with plasmids expressing HA-K27-Ub together with an EV or Myc-Stub1 expression vector, followed by VSV (MOI = 0.1) infection for various durations, were subjected to immunoprecipitation with an anti-TBK1 antibody and immunoblot analysis with the indicated antibodies. B Co-IP and immunoblot analysis of HEK293T cells transfected with Flag-TBK1 and HA-K27-Ub, together with vectors for WT Myc-Stub1 or its mutants. Protein lysates were harvested after CQ (50 μM) treatment (6 h) for immunoblotting with the indicated antibodies. C HEK293T cells were transfected with vectors for WT Myc-Stub1 or its mutants, followed by SeV (MOI = 0.1) infection for 12 h. Protein lysates were immunoprecipitated with an anti-TBK1 antibody and immunoblotted with the indicated antibodies. D Co-IP and immunoblot analysis of HEK293T-stub1+/+ and HEK293T-stub1−/− cells transfected with Flag-TBK1 and HA-K27-Ub, together with vectors for WT Myc-Stub1 or its mutants. Protein lysates were harvested after CQ (50 μM) treatment (6 h) for immunoblotting with the indicated antibodies. E Co-IP analysis of the ubiquitination of endogenous TBK1 in BMDMs from Stub1fl/fl and Stub1fl/fl Lyz2-Cre mice that were mock infected (mock) or infected with VSV for 12 h. F Analysis of the ubiquitination sites of the ULC domain of TBK1 (above). Co-IP and immunoblot analysis of protein extracts of HEK293T cells transfected with vectors expressing WT Flag-TBK1 or its mutants (K323R, K341R, K344R, K372R), HA-K27-Ub and EV or vector for Myc-Stub1, in the presence of CQ (50 μM) for 6 h (below). G HEK293T cells were transfected with plasmids expressing WT Flag-TBK1 or its indicated mutants, together with plasmids for HA-Stub1, in the presence of CQ (50 μM) for 6 h, followed by immunoprecipitation with anti-Flag beads and immunoblotting with an anti-HA antibody. H Immunoblot analysis of protein lysates of HEK293T cells transfected with HA-Stub1 together with plasmids expressing WT Flag-TBK1 or the indicated mutants. The data are representative of three independent experiments with three biological replicates (means ± SDs) or represent three independent experiments with similar results (A–H).
We overexpressed Stub1, TBK1, and a series of ubiquitin (Ub) mutants in HEK293T cells: wild-type ubiquitin (WT-Ub), K27 point mutation ubiquitin (K27R-Ub), ubiquitin lacking lysine (K0), K27-only ubiquitin (K27-only Ub), and tandem ubiquitin (Ub2, Ub4)—the latter specifically designed to mimic short K27-linked chains in HEK293T cells. The stability of the TBK1 protein was assessed in total cell lysates via Western blot analysis. TBK1 was enriched via immunoprecipitation and analyzed for K27-linked ubiquitination by immunoblotting with a K27-linked-specific antibody. The results revealed that K27-only Ub (but not the ubiquitin-deficient mutants K27R or K0) significantly enhanced Stub1-mediated TBK1 degradation and induced pronounced K27-linked polyubiquitination. Preformed K27-linked dimeric ubiquitin (Ub2) was sufficient to drive degradation, indicating that short K27-specific conjugates constitute the minimal degradation signal (Fig. S8D). The temporal relationship between TBK1 activation and ubiquitination remains to be elucidated. Bone marrow-derived macrophages were isolated from Stub1fl/fl mice and subsequently infected with VSV at multiple time points. The present study detected TBK1 phosphorylation and ubiquitination and revealed that significant phosphorylation occurred 4 h post-VSV infection. Concurrently, TBK1 K27 ubiquitination also began to increase (Fig. S8E). On the basis of these findings, we propose that TBK1 phosphorylation may alter its conformation, thereby promoting K27 ubiquitination and subsequent degradation, which could modulate TBK1-mediated signaling pathways.
Notably, Stub1 interacts with the ULD domain of TBK1. To gain insight into the mechanism by which Stub1 regulates TBK1 ubiquitination, we analyzed and identified four lysine residues (K323, K341, K344, and K372) within the TBK1 ULD domain and generated mutants ranging from lysine (K) to arginine (R) (Fig. 5F). Our results revealed that Stub1 was unable to facilitate K27-linked polyubiquitination in the TBK1 K344R mutant (Fig. 5F). Furthermore, we discovered that the TBK1 K344R mutant did not interact with Stub1 (Fig. 5G) and that Stub1 was unable to promote the degradation of the TBK1 K344R mutant (Fig. 5H). To address the functional importance of ubiquitination at the K344 site of TBK1 in type I IFN signaling, we overexpressed Stub1 with a TBK1 or TBK1 K344R mutant in TBK1 KO cells and found that Stub1 failed to inhibit TBK1-mediated IFN production after VSV or HSV-1 infection when it was restored with the TBK1 K344R mutant in TBK1 KO cells (Fig. S8F, G). Collectively, these findings suggest that Stub1 specifically promotes the K27-linked polyubiquitination of TBK1 at K344.
Stub1 promotes TBK1 degradation through chaperone-mediated autophagy
To investigate the underlying mechanisms of the regulation of TBK1 degradation by Stub1, cells co-expressing Stub1 and TBK1 were treated with different indicated inhibitors. As shown in Fig. 4J, only the lysosomal inhibitor CQ, but not the autophagosome formation inhibitor 3-MA or the autophagosome-lysosome fusion inhibitor Baf-A1, suppressed TBK1 degradation, indicating that Stub1 promotes TBK1 degradation independent of macroautophagy. In addition, analysis of the human TBK1 amino acid sequence revealed the presence of three canonical putative KFERQ-like motifs (Fig. S9A, B). To confirm which CMA motif is key to Stub1-mediated TBK1 degradation, we constructed three CMA motif mutant plasmids of TBK1 (M1-VAALA, M2-QAAKA, and M3-KAAKA) and experimentally demonstrated that the KFDKQ motif regulates Stub1-mediated TBK1 degradation by CMA (Fig. S9C).
We inferred that Stub1 may promote TBK1 degradation in a CMA-dependent manner. Lysosome-associated membrane protein 2A (LAMP2A) and HSC70 are central proteins involved in CMA. Previous data have shown that HSC70 recognizes and interacts with the CMA motif of target proteins, whereas LAMP2A, located on the lysosome membrane, facilitates the transport of target proteins into lysosomes for degradation [45]. After the overexpression of TBK1, HSC70, and Stub1 in cells, the coimmunoprecipitation results revealed that TBK1 interacted with HSC70 and that Stub1 increased their interaction (Fig. 6A). Consistently, we also noted that endogenous HSC70 interacted with TBK1 during VSV and HSV-1 infection (Fig. 6B, C). BIFC results also confirmed the interaction between HSC70 and TBK1 (Fig. 6D). To determine whether the K27 chain of TBK1 is essential for HSC70 binding, we examined the interaction between TBK1-WT and TBK1-K344R with HSC70. We found that after the K27 ubiquitin-binding site of TBK1 was mutated, TBK1 no longer interacted with HSC70 (Fig. 6E). To further demonstrate that HSC70 specifically recognizes K27-linked polyubiquitination of TBK1, we performed in vitro binding assays. The results showed that HSC70 only bound to K27-ubiquitinated TBK1, but not to K48-ubiquitinated TBK1 (Fig. S10A). Subsequently, to investigate whether other heat shock proteins are involved, we also conducted in vitro binding experiments. The results indicated that only HSC70—and not HSP70 or HSP90—specifically bound to K27-ubiquitinated TBK1 (Fig. S10B), underscoring the specificity of HSC70 in recognizing TBK1 during chaperone-mediated autophagy (CMA). We noted that the overexpression of HSC70 increased Stub1-mediated TBK1 degradation. Additionally, VER-155008, an HSC70 inhibitor that inhibits CMA [46], was used to treat cells. We observed that Stub1-regulated TBK1 degradation was greatly reduced in VER-155008-treated cells (Fig. 6F). Notably, the overexpression of HA-HSC70 and Stub1 significantly improved the degradation of TBK1 in HEK293T-hsc70−/− cells (Fig. 6G), although the knockout of HSC70 did not affect the interaction between TBK1 and Stub1 (Fig. 6H). However, Stub1 knockout significantly attenuated the interaction between TBK1 and HSC70 (Fig. 6I). In addition, compared with that in HEK293T-hsc70+/+ cells, the expression level of TBK1 in the lysosomes of HEK293T-hsc70−/− cells was significantly lower after VSV infection (Fig. 6J). Furthermore, the confocal microscopy results also revealed that TBK1 does not localize to lysosomes following VSV infection in HEK293T-hsc70−/− cells (Fig. 6K). These results suggest that Stub1 may promote TBK1 degradation through CMA.

A HEK293T cells were transfected with Flag-TBK1, Myc-Stub1, and HA-HSC70 plasmids for 24 h, and then the lysates were immunoprecipitated with an anti-Flag antibody and analyzed by immunoblotting. B, C THP-1 cells were infected with VSV or HSV-1 for the indicated times and then subjected to immunoprecipitation with a TBK1 antibody before immunoblot analysis of endogenous TBK1 and HSC70 protein levels. D BIFC analysis of TBK1 and HSC70. HA-HSC70-VN, HSC70 fused with the N-terminus of Venus; Flag-TBK1-VC, TBK1 fused with the C-terminus of Venus. E Co-IP and immunoblot analysis of HEK293T cells transfected with Flag-TBK1 or Flag-TBK1-K344R and HA-HSC70. F HEK293T cells were transfected with a Flag-TBK1 plasmid with or without HA-HSC70 plasmids and then treated with VER-155,008 (5 μM, 10 μM, or 25 μM). TBK1 protein levels were analyzed by immunoblotting. G HEK293T-hsc70−/− cells were transfected with Flag-TBK1, Myc-Stub1, and HA-HSC70 plasmids for 24 h, after which the lysates were analyzed by immunoblotting. H HEK293T-hsc70+/+ and HEK293T-hsc70−/− cells were transfected with Flag-TBK1 and HA-Stub1 plasmids for 24 h, after which the lysates were immunoprecipitated with an anti-Flag antibody and analyzed by immunoblotting in the presence of CQ (50 μM) for 6 h. I HEK293T-stub1+/+ and HEK293T-stub1−/− cells were transfected with Flag-TBK1 and HA-HSC70 plasmids for 24 h, and then the lysates were immunoprecipitated with an anti-Flag antibody and analyzed by immunoblotting in the presence of CQ (50 μM) for 6 h. J HEK293T-hsc70+/+ and HEK293T-hsc70−/− cells were infected with VSV for 0, 4, 8, or 12 h in the presence of CQ (50 μM). Lysosomes were subsequently isolated and enriched before the proteins were analyzed by immunoblotting. K Fluorescence images showing the colocalization of TBK1 (green) with HSC70 (cyan) on lysosomes (red) in hsc70+/+ cells, whereas TBK1 (green) does not colocalize with lysosomes (red) in hsc70−/− cells; original magnification ×600. The data are representative of three independent experiments with three biological replicates (means ± SDs) or represent three independent experiments with similar results (A–K).
Stub1 promotes TBK1 localization in lysosomes
The substrate-HSC70 complex has been shown to interact with LAMP2A, which mediates the translocation of the substrate into the lysosome for degradation [47]. To assess the relationships among Stub1, HSC70, and LAMP2A, these three proteins were coexpressed in HEK293T cells. We found that Stub1 and HSC70 can synergistically promote TBK1 degradation (Fig. 7A). Co-IP results indicated that Stub1 interacted with TBK1, LAMP2A, and HSC70 (Fig. 7B). To further determine whether the interaction between Lamp2a and TBK1 occurs in the cytoplasm, lysosomes, or other vesicular compartments, we isolated lysosomes and performed co-IP using lysosomal extracts to detect the interaction between TBK1 and Lamp2a. The results indicate that TBK1 and Lamp2a interact within lysosomes and that Stub1 enhances this interaction (Fig. 7C). To definitively demonstrate the specific degradation of TBK1 by CMA, we established a LAMP2A knockout cell line via CRISPR-Cas9 technology. Cotransfection analysis of TBK1 and Stub1 in LAMP2A-deficient cells revealed that Stub1-mediated degradation of TBK1 was completely inhibited (Fig. 7D). This finding further corroborates the indispensable role of CMA in this specific process. Subsequent retransfection of LAMP2A restored TBK1 degradation, thereby reactivating Stub1’s ability to promote TBK1 protein degradation. This finding suggests that the lysosomal degradation specificity of TBK1 is achieved through the CMA pathway rather than through alternative mechanisms (Fig. 7D). Confocal microscopy results further revealed that TBK1 and LAMP2A exhibited distinct colocalization within lysosomes. In the absence of LAMP2A, there was a significant reduction in TBK1 colocalization with lysosomes (Fig. 7E). To investigate whether Stub1 directly promotes TBK1 degradation via the lysosomal pathway, we performed lysosomal isolation. Under basal conditions with Stub1 overexpression, we observed only a faint TBK1 signal during lysosomal isolation, which was consistent with rapid degradation upon entry. However, when cells were treated with chloroquine (CQ, 50–100 μM) to inhibit lysosomal hydrolases, a pronounced accumulation of TBK1 within lysosomes was evident. This CQ-induced accumulation was specific to the lysosomal pathway, as it was not recapitulated by the proteasome inhibitor MG-132. Furthermore, the lysosomal accumulation of TBK1, a well-established inducer of CMA, was markedly increased upon serum starvation. Critically, this accumulation was abrogated in LAMP2A knockout cells, in which TBK1 levels in lysosomes remained low even with CQ treatment. These data collectively demonstrate that TBK1 is targeted to the lysosome for degradation in a CMA-dependent manner (Fig. 7F). To further validate this finding, we performed a LAMP2A antibody blocking assay after lysosomal isolation. The results revealed that following LAMP2A antibody-mediated blockade of lysosomes, TBK1 failed to enter the lysosomal compartment (Fig. 7G). Interestingly, we also found that TBK1 can be transported to lysosomes only when Stub1 promotes the polyubiquitination of TBK1 (Fig. 7G). Overall, these findings prove that Stub1 facilitates the entry of TBK1 for degradation in lysosomes through CMA.

A HEK293T cells were transfected with Flag-TBK1, Myc-Stub1, GFP-LAMP2A, and HA-HSC70 plasmids before analysis by immunoblotting with the indicated antibodies. B HEK293T cells were transfected with Flag-TBK1, Myc-Stub1, GFP-LAMP2A, and HA-HSC70 plasmids for 24 h, and then the lysates were immunoprecipitated with an anti-Flag antibody and analyzed by immunoblotting in the presence of CQ (50 μM) for 6 h. C Flag-vector and Flag-stub1 were overexpressed in HEK293T cells. The cells were treated with CQ for 6 h prior to total protein extraction. Lysosomes were subsequently isolated for co-IP experiments to detect the interaction between TBK1 and Lamp2a. D Immunoblot analysis of TBK1 protein levels in Lamp2a+/+ and Lamp2a−/− cells cotransfected with plasmids encoding TBK1 and Stub1. Lamp2a−/− cells were subsequently reconstituted via transfection with a LAMP2A expression plasmid. E Fluorescence images showing the colocalization of TBK1 (green) with LAMP2A (cyan) on lysosomes (red) in Lamp2a+/+ cells, whereas TBK1 (green) does not colocalize with lysosomes (red) in Lamp2a−/− cells; original magnification ×600. F Stub1 was overexpressed in Lamp2a+/+ and Lamp2a−/− cells, the cells were treated with CQ or MG132 and starved, and then, the lysosomes were isolated and enriched before the proteins were analyzed by immunoblotting. G Isolated lysosomes were preincubated with a function-blocking anti-LAMP-2A antibody or control IgG. After being washed, they were incubated with purified TBK1 protein, Stub1 protein, and recombinant ubiquitin/E1/E2 proteins in CMA reaction buffer containing an ATP-regenerating system at 37 °C. Lysosomal protease inhibitors such as leupeptin were added. Lysosomes were reisolated via centrifugation, and TBK1 uptake was analyzed by immunoblotting. The data are representative of three independent experiments with three biological replicates (means ± SDs) or represent three independent experiments with similar results (A–G).
Stub1 deficiency augments host antiviral responses
Stub1fl/fl mice and Stub1fl/fl Lyz2-Cre mice were challenged with HSV-1 and VSV via intraperitoneal injection to further define the ability of Stub1 to inhibit type І IFN production and host antiviral responses in vivo. As shown in Fig. 8A, H, Stub1fl/fl Lyz2-Cre mice presented a high level of resistance to HSV-1 infection (Fig. 8A) in terms of overall survival compared with Stub1fl/fl mice and a significantly lower mortality rate caused by VSV infection in these mice (Fig. 8H). The ELISA results indicated that IFN-β and IL-6 production in the sera of Stub1fl/fl Lyz2-Cre mice was significantly greater than that in the sera of Stub1fl/fl mice (Fig. 8B, C, I, J). Consistently, we found that the mRNA levels of Ifnβ1 in the brains of Stub1fl/fl Lyz2-Cre mice were significantly greater than those in the brains of Stub1fl/fl mice after infection with HSV-1 for 48 or 72 h (Fig. 8D). Similarly, the mRNA levels of Ifnβ1 in the lungs of Stub1fl/fl Lyz2-Cre mice were significantly greater than those in the lungs of Stub fl/fl mice after infection with VSV for 48 and 72 h (Fig. 8K).

A Survival of Stub1fl/fl and Stub1fl/fl Lyz2-Cre mice treated with HSV-1 (2 × 108 PFU/g) via intraperitoneal injection (n = 10 per group). ELISA analysis of IFN-β (B) and IL-6 (C) production in sera from Stub1fl/fl and Stub1fl/fl Lyz2-Cre mice (n = 3 per group). D qPCR analysis of Ifnb1 mRNA expression in the brains of Stub1fl/fl and Stub1fl/fl Lyz2-Cre mice after VSV administration (2 × 108 PFU/g) via intraperitoneal injection (n = 3 per group) for 48 and 72 h. E qPCR analysis of HSV-1 genomic DNA expression in the brains of Stub1 fl/fl and Stub1fl/fl Lyz2-Cre mice after HSV-1 administration (2 × 108 PFU/g) via intraperitoneal injection (n = 3 per group) for 48 and 72 h. F Determination of HSV-1 loads in the brain of Stub1 fl/fl and Stub1 fl/fl Lyz2-Cre mice via the TCID50 assay. G Hematoxylin and eosin-stained images of brain sections from Stub1 fl/fl and Stub1 fl/fl Lyz2-Cre mice infected with HSV-1 for 72 h. H Survival of Stub1fl/fl and Stub1fl/fl Lyz2-Cre mice administered VSV (1 × 108 PFU/g) via intraperitoneal injection (n = 10 per group). ELISA analysis of IFN-β (I) and IL-6 (J) production in sera from Stub1 fl/fl and Stub1 fl/fl Lyz2-Cre mice (n = 3 per group). K qPCR analysis of Ifnb1 mRNA expression in the brains of Stub1 fl/fl and Stub1 fl/fl Lyz2-Cre mice after VSV administration (1 × 108 PFU/g) via intraperitoneal injection (n = 3 per group) for 48 and 72 h. L qPCR analysis of VSV genomic mRNA expression in lungs from Stub1 fl/fl and Stub1 fl/fl Lyz2-Cre mice after VSV administration (1 × 108 PFU/g) via intraperitoneal injection (n = 3 per group) for 48 and 72 h. M Determination of VSV loads in the lungs of Stub1 fl/fl and Stub1 fl/fl Lyz2-Cre mice via the TCID50 assay. N Hematoxylin and eosin-stained images of lung sections from Stub1 fl/fl and Stub1 fl/fl Lyz2-Cre mice infected with VSV for 72 h. O Scoring is based on the proportion of neutrophils among all cells: 0 points: Neutrophils absent; 1 point: Neutrophil count <5%; 2 points: Neutrophil count 5–50%; 3 points: Neutrophil count >50%. The data are presented as the means ± SDs of three independent experiments. Statistical significance was determined by two-tailed Student’s t test or one-way ANOVA followed by the Bonferroni post hoc correction, as appropriate. *P < 0.05, **P < 0.01, ***P < 0.001.
The HSV-1 DNA copy numbers and titers in the brains of Stub1fl/fl Lyz2-Cre mice were significantly lower than those in Stub1fl/fl mice (Fig. 8E, F), and the VSV copy numbers and titers in the lungs of Stub1fl/fl Lyz2-Cre mice were significantly lower than those in Stub1fl/fl mice after infection with VSV for 48 h and 72 h (Fig. 7L, M). We also observed less inflammatory cell infiltration and slight tissue injury in the brains of Stub1fl/fl Lyz2-Cre HSV-1 mice than in those of Stub1fl/fl mice after infection with HSV-1 (Fig. 8G). Fewer signs of severe inflammation and less pathological damage were observed in the lungs of Stub1fl/fl Lyz2-Cre mice than in those of Stub1fl/fl mice (Fig. 8N). Immune infiltration was scored on the basis of the proportion of neutrophils among total cells: 0 points: absence of neutrophils; 1 point: neutrophil count <5%; 2 points: neutrophil count 5%-50%; 3 points: neutrophil count >50%. The results are shown in Fig. 8O. Together, these results suggest that Stub1 suppresses IFN-I production and inhibits the antiviral innate response in the context of VSV and HSV-1 infections in vivo.