
Human samples
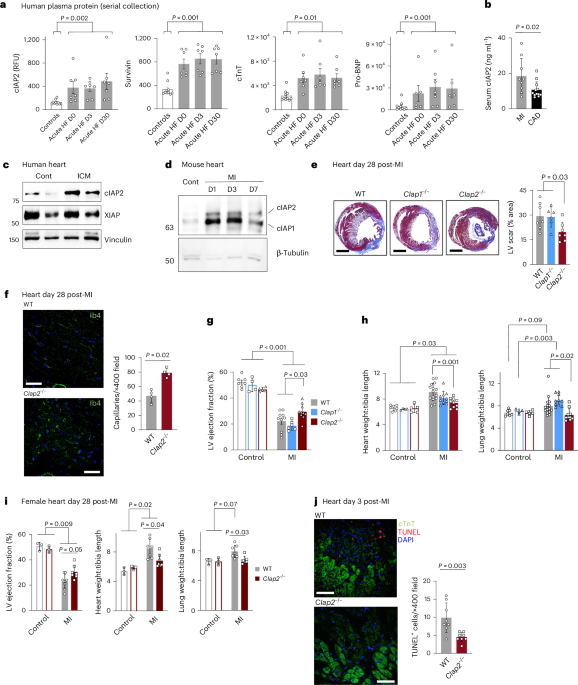
Patient samples (blood whole plasma, peripheral blood mononuclear cells (PBMCs) and whole unfractionated leukocyte lysates) were obtained with consent and used under approval from the Ottawa Health Science Network Research Ethics Board (OHSN-REB protocols no. 20140869, no. 20150428 and no. 20160516-01H). Patient characteristics are summarized in Supplementary Table 1. For whole plasma cell-free protein analysis, samples were isolated from N = 7 randomized patients presenting with acute heart failure at the time of admission and subsequently at days 3 and 30 after admission and compared with N = 10 healthy controls. Analytes were screened using a SomaScan aptamer-tagged proteomics array (SomaLogic) as described previously78. Plasma was frozen until analysis at −80 °C. Acute MI versus CAD PBMC analyses were conducted using isolates from patients admitted to the University of Ottawa Heart Institute percutaneous coronary intervention clinic, where approximately 30 ml of whole blood samples was obtained within 2 hours of admission (for planned CAD intervention, N = 12) or diagnosis of ST-segment elevated MI (STEMI, referred to as ‘MI’, N = 9). PBMC isolates were performed by gradient centrifugation of PBS-diluted whole blood over a 2:1 (v/v) Percoll gradient at 675g for 30 minutes at room temperature, followed by concentration of buffy coat cell content in RPMI 1640 + 5% (v/v) FBS (Thermo Fisher Scientific) to a final concentration of 5 × 106 cells per milliliter. Standardized quantities of cells (1 × 106 per milliliter per sample) were reserved for flow cytometric analysis. For collection of cell lysate from unfractionated leukocytes, patient samples were collected at the time of clinical assessment, followed by removal of red blood cells using hypotonic ammonium chloride/potassium bicarbonate buffer and subsequent lysis of nucleated cell pellets. Lysates were stored for use in immunoassays. Patient sera were collected from patients with MI versus patients with CAD by centrifugation of 2 ml of whole blood at 1,000g at room temperature, followed by extraction of aqueous (non-packed hematocrit) fraction. Sera were stored for analysis at −80 °C.
Mouse post-MI gene set enrichment analysis
Left ventricular expression of gene products over a 48-hour timecourse was catalogued in the National Center for Biotechnology Information (NCBI)-curated, open-source dataset GSE4648 (GDS2329) (for reference, see https://www.ncbi.nlm.nih.gov/sites/GDSbrowser?acc=GDS2329). The dataset was uploaded onto the Salk Institute open-access R Shiny bioinformatics platform BART (Bioinformatics Array Research Tool)79, and data were organized and catalogued using applications encoded in the software package (N = 2 per MI timepoint in infarcted left ventricular region or N = 2 for controls at representative timepoints). Data for differential gene expression and Gene Ontology annotation were collected from the 24-hour post-MI timepoint, and the summary of differential genes is located in Supplementary Tables 2 and 3. Statistical analyses were conducted within the software application.
Animals
C57Bl/6 (WT), Ciap2−/−, Ciap1−/−, Nod1−/−, Tlr4−/− and Tnfrsf1a−/− mice were bred and co-housed for multiple generations under identical specific pathogen-free conditions (including consistent chow, water and air supply) in the University of Ottawa Heart Institute vivarium prior to experimental procedures. Prior to breeding and use in this study, the principal experimental mouse strains Ciap2−/− and Ciap1−/− were similarly normalized to C57Bl/6 background by backcrossing as described in ref. 27 and ref. 33, respectively. Bl/6 CD45.1 mice (B6.SJL-Ptprca Pepcb/BoyJ) were purchased from The Jackson Laboratory. Mice were 12−16 weeks of age prior to surgical experiments. Mice were kept on a consistent 6:00−20:00 light/dark cycle, at constant 22° C temperature and 40% humidity and were provided chow and drinking water ad libitum. Both male and female mice were used for experimental MI procedures for germline knockout CIap2−/− studies; otherwise, males were used in reported experimental work. In all experiments, groups of animals were operated on in random order, and assignment to either experimental MI or control surgery was conducted in a blinded fashion. All animal experimentation was conducted under the guidance and with approval of the University of Ottawa Animal Care Committee (ACC), with experimental work adhering to ACC animal usage protocols HIb-2218, HIb-3536, HIe-2246 and HIe-3727. All surgical procedures were carried out with strict adherence to university-mandated protocols, including perioperative and postoperative analgesia and wellness assessment. Experiments were conceived and conducted to ensure compliance with directives from the Canadian Council for Animal Care and the ARRIVE criteria for responsible animal research.
MI
Male or female mice, aged 12−16 weeks, were randomly assigned to MI or sham (control) surgery. One hour prior to surgical procedure, mice were injected subcutaneously with a 1.2 mg kg−1 solution of buprenorphine slow-release formulation and a 1.0 mg kg−1 solution of meloxicam. Mice were anesthetized with 2−4% isoflurane (mixed with O2); hair on the left chest was removed with a depilatory cream; and mice were intubated with a 20-gauge soft catheter attached to a ventilator operating at 130−150 strokes per minute. Once in adequate anesthetic surgical plane, the chest was opened on the left rib cage between either the 4th or 5th intercostal space; the pericardium was disrupted; and the left anterior descending artery (LAD) was located and ligated with a 7-0 silk suture approximately 2 mm below the atrial appendage. Arterial occlusion was verified by blanching of the distal myocardial tissue, at which time the chest was closed with a 6-0 Surgipro polyvinyl suture. Sham-operated controls underwent the same procedure foregoing the terminal LAD ligation. The animals were allowed to recover from anesthetic in a 30 °C hyperbaric incubator and received 1.0 mg kg−1 meloxicam daily for two subsequent days. Food and water were not withdrawn at any point prior to or after surgery. Mice were subsequently monitored for the duration of the experimental protocol, determined at random to last 1, 3, 7 or 28 days unless endpoint was determined by wellness assessment.
Isoproterenol treatment
Twelve- to 16-week-old male C57Bl/6 mice were injected subcutaneously (subscapular) with 10 mg kg−1 isoproterenol-HCl in saline. Twenty-eight days after injection, cells and protein lysates were isolated and used in flow cytometry or immunoblotting assays.
Blood, spleen, bone marrow and heart immune cell isolation
PBMCs
Whole blood (50−100 μl) was collected by temporal vein lancet puncture and lysed with 20 volumes (1−1.5 ml) of cold red blood cell lysis buffer (150 mM ammonium chloride (NH4Cl), 20 mM potassium carbonate (K2HCO3) and 0.1 mM EDTA). Suspensions were centrifuged at 330g, and non-lysed cell pellets were resuspended in either PBS supplemented with 1% BSA for flow cytometry analyses or cell lysis buffer (PBS supplemented with 1% (v/v) Triton X-100, 0.25% (v/v) sodium deoxycholate, 5 mM EDTA, 10 mM sodium orthovanadate, 2 mM sodium fluoride and 1× concentrated cOmplete protease inhibitor cocktail).
Spleens
Whole spleens from sham or MI-treated mice were excised, washed in cold sterile PBS and disrupted by crushing through 40-μm nylon mesh strainers. Cells were then centrifuged at 330g and resuspended in red blood cell lysis buffer. Non-lysed cells were recentrifuged at 330g, and cell pellets were resuspended in PBS plus 1% BSA (for flow cytometry), RPMI 1640 + 5% FBS (for adoptive transfer) or cell lysis buffer (for immunoblot protein analyses).
Bone marrow
Long bones (either tibia or femurs) were excised; muscle and connective tissues were removed; and bones were washed in cold 70% ethanol and PBS, consecutively. Bones were then crushed using a mortar and pestle. Marrow was resuspended in cold PBS and passed through a 40-μm nylon strainer, and cell suspensions were centrifuged at 330g, followed by resuspension in PBS/1% BSA, RPMI 1640 + 5% FBS or cell lysis buffer as above.
Hearts
Hearts were excised and flushed with cold PBS, and left ventricular tissue was dissected. In preliminary experiments where both MI or isoproterenol treatments were evaluated, the apical (approximate lower third) region was removed, and cells were isolated; in focused MI experiments, the infarct-affected (border) region, which we defined as the approximate 2-mm boundary on either side of the infarcted region (identified by fibrosis or blanching of the myocardium), was removed and used for cell isolation and flow cytometry or for whole-tissue molecular analyses (RT−qPCR or immunoblotting; Supplementary Fig. 1). Left ventricular tissue extracts were weighed and digested in tissue digestion buffer (113 mM NaCl, 0.6 mM KH2PO4, 0.6 mM Na2HPO4, 20 mM HEPES, 20 mM NaHC03, 4.7 mM KCl, 2 mM MgCl2, 1 mM EDTA, 60 U ml−1 DNAse I (MilliporeSigma), 100 U ml−1 hyaluronidase (MilliporeSigma), 450 U ml−1 Collagenase I (Worthington Biochemical), 250 U ml−1 Collagenase II (Worthington Biochemical) and 125 U ml−1 Collagenase D (Worthington Biochemical)) by shaking at 37 °C for 1 hour. Liberated cells were gravity filtered through a 40-μm nylon strainer by washing with cold PBS. Cell suspensions were centrifuged at 330g and resuspended in either PBS/1% BSA or RPMI 1640/5% FBS. For quantitation purposes, aliquots of all cell suspensions (prior to red blood cell lysis or centrifugation) were collected; 1 in 10 dilutions were made in PBS; and cells were counted using a hemocytometer.
Flow cytometry and fluorescence-activated cell sorting
Cell isolates collected as described above were stained with specified antibody cocktails at a concentration of approximately 2.5 × 106 cells per milliliter in PBS supplemented with 1% BSA for 90 minutes on ice protected from light. Antibody concentrations were prepared as master cocktail mixes with specific antibodies employed at the concentrations indicated in Supplementary Table 4. For cytometric analysis, stained cells were washed with PBS, spun down at 330g and resuspended in PBS supplemented with 1% v/v paraformaldehyde. Stained cells were either immediately analyzed or stored overnight at 4 °C protected from light. Gating strategies for cytometry experiments are provided in the Supplementary Data file. For fluorescence-activated cell sorting (FACS) purification and cell recovery, stained cell isolates were washed, resuspended in RPMI 1640/5% FBS and immediately resolved. Cells were collected into chilled 5-ml round-bottom polypropylene tubes containing 1 ml of RPMI 1640/5% FBS. Where possible, cell purifications were verified by performing reanalysis on aliquots of recovered sample or by microscopic assessment on a hemocytometer. All cytometric processing was carried out on a FACSAriaIII four-laser cell sorter/cytometer (BD Biosciences). Experiments were acquired using BD FACSDiva version 8.2 software, and analysis was performed using either FACSDiva or FlowJo version 10.8 software (Tree Star).
Bone marrow transplantation
Bone marrow chimeric mice were established by lethally irradiatiating 6−8-week-old B6 CD45.1 mice with exposure to two cycles (spaced by 20-minute intervals) of 450-cGy gamma irradiation using a Gammacell 3000 Cesium137 source irradiator (Best Theratronics). Within 3 hours of irradiation, mice were injected intravenously with 2.5 × 106 sterile-isolated, filtered bone marrow cells from C57Bl6/, CIap2−/− or CIap1−/− donors. Mice were allowed to recover over a minimum 6-week period in a sterilized housing unit, after which a blood sample was collected to assess hematopoietic reconstitution by flow cytometric analysis of CD45.1 (recipient strain) and CD45.2 (donor strain) isotypes as well as verification of PBMC populations. Chimerism of 90% or greater was consistently achieved, with no apparent deficiency in specific cell populations. Animals were subsequently randomized into MI experiments, with sample groups aggregated from three separate similar experimental protocols. In all experiments, small groups of mice were randomly assigned to sham surgical procedure. For ‘reciprocal’ bone marrow transplantation experiments, CD45.2 (C57Bl/6 or CIap2−/−) mice were irradiated and served as recipient strains for CD45.1 bone marrow donation in order to assess the role of resident cardiac cIAP2 expression after injury. Experiments were presented as aggregate data from two similar experimental protocols.
Antibody injections for in vivo neutralization studies
TRAIL (Tnfsf10) neutralization using N2B2 monoclonal antibody
C57Bl/6 or CIap2−/− mice (as above) were injected with either 250 μg of rat anti-TRAIL IgG2a(κ) monoclonal antibody (clone N2B2; BioLegend, 109308) or equivalent quantity of isotype control rat IgG2a (clone RTK2758; BioLegend, 400502) via intraperitoneal injection 3 days before surgery, perioperatively or 3 days after surgery. Apoptosis in spleens of MI-operated mice was evaluated in a subgroup of animals at 3 days after MI, and the remaining animals were euthanized at day 28 for assay of cell infiltration or cardiac injury, as indicated in Fig. 3j,k and Extended Data Fig. 7e,f.
Smac mimetic treatments
The monovalent Smac mimetic LCL161 was purchased from Selleck Chemicals. For in vitro experiments, LCL161 was dissolved in dimethylsulphoxide (DMSO) and used at 5-μM concentrations applied to cultured bone marrow-derived dendritic cells (described below) for 48 hours in the presence or absence of inflammatory stimulation. Control cultures were treated with DMSO only. For in vivo experiments, mice were first weighed and then administered 10 mg kg−1 LCL161 dissolved in sodium acetate buffer (0.07 M NaOAc, 0.03 M HCl (pH 4.6)) orally by intragastric gavage at day 1 or days 1 and 4 after MI. Control mice were gavaged with acetate buffer only. Animals were monitored for up to 7 days or 28 days after MI, as indicated.
Echocardiography
Mice were sedated with 1.5% isoflurane (per liter O2), and hair was removed from the left chest. Parasternal long axial (high resolution, electrocardiogram-gated kilohertz visualization (EKV) and low resolution, B mode) and short axial (M mode) images were collected using a Vevo 770 or a Vevo 3100 ultrasound imaging station (FUJIFILM/VisualSonics). Systolic function (ejection fraction and stroke volume) was estimated by the observed change in two-dimensional area measurements of endocardial wall from end-diastole to end-systole using parasternal long axis (planar projection of whole ventricle from aortic valve to apex) high-resolution (300 images per beat) EKV image analysis; dimensional change and myocardial wall thickness were estimated by linear measurement of midventricular chamber from anterior to posterior wall in short axial (vertical projection) images. Imaging was performed by a blinded operator, and image analysis was carried out in random order by a blinded investigator. Measurement and calculation of imaging results was performed with Vevo LAB software (version 3.0-770 or version 3.1.1).
Histology
Hearts were excised, flushed with cold PBS and immersed in 0.5 M KCl for 5 minutes to enhance ventricular dilatation. Subsequently, hearts were fixed in 20 volumes (approximately 3 ml for mouse hearts) of 4% paraformaldehyde for 24−48 hours, reimmersed in PBS for another 48 hours and then equilibrated in autoclaved water and stored at 4 °C until being embedded in paraffin. For tissue sectioning, full thickness (concentric) 5-μm sections were cut using a microtome and collected onto slides. Tissue sections were deparaffinized using toluene and rehydrated by immersion in stepwise decreasing ethanol gradients and then subjected to hematoxylin and eosin staining, Masson’s trichrome or immunofluorescence microscopy. Midventricular sections corresponding to regions spanning the infarction were selected (based upon presence of silk suture fibers from LAD ligation; approximately similar tissue regions were selected from sham-operated mice) and used for analysis.
Tissue morphometry
Serial midventricular sections from MI-treated or sham-operated mouse hearts were stained with Masson’s trichrome (to expose collagenous scar tissue, stained blue), and collagen area was estimated as a fraction of whole left ventricle by blinded measurement using ImageJ (version 3) open-source software (National Institutes of Health). Mean area calculations from each subject were used to estimate scar area, where a minimum of five animals per MI treatment group were used for measurement.
Cell culture experiments
Reagents were purchased from Thermo Fisher Scientific unless otherwise indicated. Isoproterenol-HCl was purchased from MilliporeSigma. iE-DAP and poly I:C were purchased from InvivoGen. Flt3 ligand (Flt3L) was purchased from R&D Systems/Bio-Techne. Bone marrow-derived plasmacytoid dendritic cells: Whole bone marrow was isolated from long bones as described, and 2 × 106 cells per milliliter were seeded into 12-well culture dishes (500 ml per well) with RPMI/5% FBS supplemented with 50 ng ml−1 Flt3L and re-fed every third day for 7 days. The final 3 days, cells were additionally administered 5 µM poly I:C to further mature the cell populations80. Cells were subsequently stimulated with 10 μM ie-DAP and 20 μg necrotic heart tissue. Following 48 hours, cultured cell lysates were collected and immunoblotting was performed for proteins as presented in as presented in Extended Data Fig. 8b.
Immunofluorescence microscopy
Formalin fixed, paraffin-embedded heart midventricular sections were deparaffinized, and antigen retrieval was performed by either a 15-minute incubation in 1× proteinase K at 37 °C (for TUNEL staining or intracellular antigen staining) or a 10-minute steaming at 65 °C with 10 mM sodium citrate buffer (for surface antigen staining or isolectin B4 staining). TUNEL assay was conducted using a Click-iT EdU kit (AlexaFluor 594) (Thermo Fisher Scientific) according to the manufacturer’s specifications with nuclear counterstaining using DAPI. For immunostaining, sections were blocked for 30 minutes with 2% normal goat or donkey serum (see below) in PBS. Primary antibodies (Supplementary Table 5) were incubated with sections using indicated dilutions overnight at 4 °C, followed by washing with PBS plus 0.05% (v/v) Tween 20 detergent. Secondary antibodies (goat anti-mouse or donkey anti-rabbit) conjugated to various AlexaFluor fluorochromes were used at reported dilutions in the appropriate blocking buffer (determined by host species) and incubated on sections at room temperature for 90 minutes, followed by washing with PBS 0.05%/Tween 20 and PBS, sequentially. Staining of myocardial vasculature was performed using Griffonia simplicifolia isolectin B4 conjugated with AlexaFluor 488 (Thermo Fisher Scientific) at a concentration of 1μg ml−1 (diluted in 10 mM MgCl2/MnCl2/CaCl2) overnight at 4 °C and counterstained with DAPI. Slides were mounted with ProLong Antifade Gold (Thermo Fisher Scientific). The photomicrographs were captured in a blinded fashion using an Olympus IX81 laser confocal microscope, and images were prepared with Fluoview version 4.3 software (Olympus Corporation). Quantitation of analytes or antigens was conducted by counting events per high-powered field, minimum four fields per ventricle or splenic section, using at least triplicate animals per treatment. For high-resolution micrographs, slides were imaged on a Zeiss ELYRA LSM 880 microscope using a ×20 planar apochromat objective with AiryScan function. Images were acquired and processed using Zeiss ZEN 2.3 software (Carl Zeiss) and Imaris Viewer (version 9.8.0, Oxford Instruments) (for z-stack deconvolution).
Protein extracts from fresh-frozen tissue
Border regions of post-MI left ventricular tissue and spleens were excised at predetermined timepoints after the experimental procedure and washed in cold PBS. After being patted dry on a sterile filter paper and then weighed, tissues were placed in 2.0-ml screwcap cryovials and immersed in liquid nitrogen for storage at −80 °C. For protein isolation, tissues were thawed on ice in cold lysis buffer, finely minced with scissors and then disrupted using a glass homogenizer. Suspensions were incubated on ice for 1 hour with occasional vortexing and then sonicated twice for 10-second intervals. Supernatants were clarified by centrifugation at 12,000g at 4 °C for 10 minutes and transferred to new sample tubes for use in immunoblotting. Prior to electrophoresis, protein quantities were determined by Bradford assay.
Nuclear extracts
Halved spleens (approximately 35−40 mg), either freshly isolated or frozen, were incubated in 1.5-ml Eppendorf tubes on ice in cold PBS followed by PBS diluted 1:5 in distilled deionized water. Tissue was minced into small pieces with scissors and spun at 500g, and PBS solution was replaced with hypotonic lysis buffer (10 mM HEPES (pH 7.5), 10 mM KCl, 0.1 mM EDTA, 1 mM DTT, 1× concentrate cOmplete protease inhibitor cocktail and 50 mM sodium orthovanadate). Tissue was further minced, vortexed gently and incubated for 10 minutes on ice, followed by addition of 0.5% Triton X-100. Cell suspensions were spun at 1,500g; supernatants (‘cytoplasmic’ fraction) were transferred to collection tubes; and pellets were resuspended in nuclear extraction buffer (20 mM HEPES (pH 7.5), 20 mM KCl, 80 mM NaCl, 25% (v/v) glycerol, 0.5 mM EDTA, 1 mM DTT, 1× concentrate cOmplete protease inhibitor cocktail and 50 mM sodium orthovanadate) for 30 minutes on ice. Suspensions were intermittently vortexed and finally centrifuged at 12,000g (4 °C) for 15 minutes. Supernatants were collected, and both cytoplasmic and nuclear extracts were stored at −80 °C for use in immunoblotting. Prior to electrophoresis, sample protein concentration was measured by Bradford assay.
Immunoprecipitation and immunoblotting
Immunoprecipitation
300 μg of cell extracts was incubated in lysis buffer and 2 μg ml−1 mouse monoclonal anti-TRAF3 (Santa Cruz Biotechnology), rabbit ployclonal anti-TRAF6 (Cell Signaling Technology) or mouse monoclonal anti-RIPK1 (BD Biosciences) antibodies, respectively. Samples were rotated continuously at 4 °C overnight and subsequently incubated with 0.10 volumes of Protein G-conjugated Dynabeads (Thermo Fisher Scientific) at 4 °C for an additional 90 minutes. Antigen-antibody complexes were immobilized using a DynaSpin column (Thermo Fisher Scientific), and the unbound fraction was saved for immunoblot analysis. Beads were subsequently washed in lysis buffer and PBS, sequentially, and antigen-antibody complexes were eluted using 4× Laemmli sample loading buffer and heating at 95 °C for 5 minutes. Samples were loaded onto 8% SDS-PAGE gels, and isolated proteins were subsequently probed by immunoblotting for proteins as indicated. Ubiquitin pulldowns were performed using a Pierce Ubiquitin Enrichment Kit (Thermo Fisher Scientific) according to the manufacturer’s protocol, and eluted proteins were stored at −80 °C prior to immunoblotting.
Immunoblotting
Protein lysates collected as described were loaded as 10-μg aliquots onto 6% or 12% SDS-PAGE gels. Separated proteins were transferred onto polyvinyl difluoride (PVDF) membranes and immunoblotted with primary antibodies as indicated (listed in Supplementary Table 6). HRP-conjugated secondary antibodies (1:25,000 dilution; Bio-Rad) were incubated with blots, and labeled antibody-antigen complexes were developed using West Pico PLUS ECL reagent (Thermo Fisher Scientific) according to the manufacturer’s protocol. Blots were exposed and images collected on a Bio-Rad ChemiDoc XRS+ imaging system, and analysis was conducted using Image Lab version 6 software.
RNA isolation and real-time qPCR
Frozen peri-infarct region left ventricular tissue or spleen (approximately 40−60 mg) was digested in 800 µl of TRIzol solution (Thermo Fisher Scientific), followed by standard chloroform separation and 75% ethanol RNA precipitation. RNA concentrations were determined with a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific), and 1 μg of RNA was reverse transcribed to cDNA using All-In-One 5× RT PCR MasterMix (Applied Biological Materials) according to the manufacturer’s protocol. Sample cDNA was then diluted 1:10 and used for qPCR gene expression analysis using BrightGreen 2× qPCR MasterMix (Applied Biological Materials), with 10 mM target-selective primers as listed in Supplementary Table 7. PCR reactions were performed using the following cycling conditions: 95 °C for 15 seconds, 60 °C for 15 seconds and 72 °C for 15 seconds over a 40-cycle reaction sequence on a Roche LightCycler 96 instrument. Quantitation of transcript was performed using the ΔΔCt method, using HPRT or β-actin (indicated in the figure legends) as reference (control) PCR products. For most experiments, ΔΔCt values (the effect of MI on the target gene of interest) were compared between WT and CIap2−/− mice, and a relative expression ratio was calculated with WT MI expression change being assigned a reference value of 1.
ELISA
For clinical serum or lysate samples, proteins were isolated as described above and were used for measurement by human cIAP1 or cIAP2 ELISA (MilliporeSigma, RAB1369 and RAB0027). Assays were conducted as recommended according to the manufacturer’s protocols. Intracellular-isolated cIAP measurements (from PBMC lysates) were normalized by standard Bradford protein assay. For mouse analyses, spleen cells were cultured at densities of 1 × 106 cells per milliliter in RPMI/5% FBS for 48 hours, and conditioned media were collected and stored at −80 °C for use in ELISA. Conditioned media from splenocyte cultures were collected 48 hours after cell plating of 1 × 106 cells per well (12-mm wells) and subsequently used for detection of IL-10 or IL-23 using DuoSet ELISA kits (IL-10: DY417; IL-23: DY1887 (R&D Systems/Bio-Techne)) according to the manufacturer’s protocols.
Statistical analysis
Sample size calculations were performed for mouse MI experiments based upon estimation of effect sizes from preliminary experiments. In general, observation of relative performance (that is, magnitude of biological effect size) was compared among WT, CIap2−/− or CIap1−/− mice. Results were applied to subsequent experiments, with sample sizes determined according to methods summarized in ref. 81 using calculation formulas suitable for continuous variable measurements. A single exception was the use of linear regression statistical testing for survival analysis in Fig. 5d, where subject animal differences were evaluated ad hoc during experiments for cardiac injury measurements. Statistical calculations for experimental outputs were made from individual raw data using GraphPad Prism version 8 or version 9 software (GraphPad Software); sample sizes and selection of statistical tests are indicated in the figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.