
Expression and purification of S antigens
DNA constructs encoding MERS-CoV England-1 S with the 2 P substitutions45, NL140422 S with 2 P and A885P substitutions25, and HKU4 SM3A S with 2 P and T985P substitutions were synthesized (Gene Universal, Newark, DE). Each sequence was fused to a T4 fibritin trimerization domain, AviTag, and C-terminal 6×His Tag, and cloned into pcDNA3.1(-). Expi293F cells (RRID: CVCL_D615) were transfected with the plasmids per manufacturer protocol via the ExpiFectamine Transfection Kit (Thermo Fisher Scientific). Supernatants were harvested 5 days post-transfection by centrifugation (6000 × g, 10 min), followed by two rounds of dialysis against 1×PBS (2 h each, with fresh buffer). Dialyzed supernatants were incubated overnight at 4 °C with HisPure Ni-NTA resin (1 mL, Thermo Fisher Scientific) under gentle stirring. The resin–protein mixture was loaded onto a gravity flow column (G-Biosciences), washed with 90 mL of binding buffer (150 mM Tris, 150 mM NaCl, 20 mM imidazole, pH 8), and eluted in three fractions using 3 mL of elution buffer (150 mM Tris, 150 mM NaCl, 400 mM imidazole, pH 8).
Eluates were concentrated to ~1 mL using a 10 kDa MWCO centrifugal filter (Millipore Sigma) and further purified by size-exclusion chromatography on a Superdex 200 Increase 10/300 column (Cytiva) equilibrated with PBS, monitored through absorbance at 280 nm, as described previously32,33,34,35,36. Protein concentrations were determined by bicinchoninic acid (BCA) assay (Thermo Scientific), where dosing is based on the amount of S included in each sample.
Expression and purification of MS2
The gene encoding a single-chain MS2 coat protein dimer containing an AviTag insertion between residues 14 and 15 of the second monomer33 was synthesized and cloned into pET-28b (GenScript Biotech, Piscataway, NJ). The plasmid was co-transformed with pAcm-BirA (Avidity LLC) into BL21(DE3) E. coli (New England Biolabs) following the manufacturer’s protocol. Transformed cells were grown overnight at 37 °C in 5 mL of 2xYT medium and subsequently scaled to a 1 L culture. Cells were cultured at 37 °C with shaking at 225 rpm until reaching an OD600 of 0.6, at which point protein expression was induced with 1 mM IPTG (Fisher BioReagents) and supplemented with 50 nM D-biotin. Cultures were then shifted to 30 °C and incubated overnight. Cells were harvested by centrifugation (7000 × g, 7 min), and the pellet was resuspended in 25 mL lysis buffer (20 mM Tris, 0.5 mg/mL lysozyme, 125 units of EMD Millipore benzonase, one-quarter SigmaFast EDTA-free protease inhibitor tablet; pH 8). Suspensions were incubated on ice for 20 min, followed by addition of 0.1% (w/v) sodium deoxycholate. Cell disruption was performed by sonication (35% amplitude, 3 s pulses, 3 min) twice with a 5 min interval. Lysates were clarified by centrifugation (19,000 × g, 30 min), and the supernatant was diluted threefold before purification. Clarified lysates were passed through four HiScreen Capto Core 700 columns (Cytiva) connected in series and washed with five column volumes of 20 mM Tris (pH 8). The eluate was concentrated using a 10 kDa MWCO centrifugal filter and further purified by size-exclusion chromatography on a Superdex 200 Increase 10/300 column (Cytiva) equilibrated in TBS (20 mM Tris, 20 mM NaCl, pH 8). Final protein concentrations were determined by BCA assay (Thermo Fisher Scientific).
In vitro biotinylation of AviTagged S and MS2 proteins
The AviTag on MS2 and S protein was biotinylated using the BirA-500 kit (Avidity LLC) per manufacturer protocol. Briefly, 45 μM AviTagged protein was incubated with BirA ligase and Biomix B (biotin, ATP, and magnesium acetate) overnight at 4 °C. The same amount of Biomix B was added twice with a 2 h incubation at 37 °C in between and incubated overnight at 4 °C. The next morning, the mixture was run on a Superdex 200 Increase 10/300 column (Cytiva) to remove BirA and excess reagents. The concentration of biotinylated S and MS2 protein was measured by BCA assay.
Expression, refolding, and purification of streptavidin
The expression and purification of streptavidin (SA) followed established methods32,33,34,35,36,46. The SA gene (Addgene plasmid #46367, a gift from Mark Howarth) was transformed into BL21(DE3) cells. Four overnight starter cultures (5 mL, 2xYT) were scaled into 1 L 2xYT each and grown at 37 °C while shaking until OD600 reached 0.6. Expression was induced with 1 mM IPTG, and cultures were incubated overnight at 30 °C. Cells were harvested (7000 × g, 7 min) and resuspended in lysis buffer (50 mM Tris, 100 mM NaCl, pH 8.0) containing 1 mg/mL lysozyme (Alfa Aesar) and benzonase (500 units; EMD Millipore). After a 30 min incubation at 4 °C, lysis was performed by sonication (35% amplitude, 3 min, 3 s on, 3 s off) in the presence of 0.1% (w/v) sodium deoxycholate. The lysis process was repeated, but with benzonase excluded. The inclusion body pellets were homogenized, sonicated (35% amplitude, 30 s), and centrifuged (27,000 × g, 15 min) three times in 100 mL of washing buffer #1 (50 mM Tris, 100 mM NaCl, 100 mM EDTA, 0.5% (v/v) Triton X-100, pH 8.0). The pellet was then homogenized, sonicated (35% amplitude, 30 s), and centrifuged (15,000 × g, 15 min) three more times in 50 mL of washing buffer #2 (50 mM Tris, 10 mM EDTA, pH 8.0). Inclusion body pellets were solubilized in resuspension buffer containing 7.1 M guanidine hydrochloride, and the solution was incubated for an hour at room temperature. After centrifugation (12,000 × g, 12 min), the supernatant was transferred to a syringe and then added dropwise at a rate of 30 mL/hour to chilled 1 L PBS with vigorous stirring. After stirring the solution overnight at 4 °C, insoluble contaminants were removed by centrifugation (17,000 × g, 15 min) and the supernatant was filtered through a 0.45 μm bottle-top filter. Next, ammonium sulfate was slowly added to the rapidly stirring supernatant to a concentration of 1.9 M and mixed at 4 °C for 3 h to precipitate out impurities. After another round of centrifugation (17,000 × g, 15 min) and filtration, more ammonium sulfate was added to the supernatant up to a final concentration of 3.68 M. After overnight stirring at 4 °C, the solution was centrifuged (17,000 × g, 15 min) to pellet out SA. Pelleted SA was resuspended in 20 mL of Iminobiotin Affinity Chromatography (IBAC) binding buffer (50 mM sodium borate, 300 mM NaCl, pH 11.0) and purified on iminobiotin agarose resin (Thermo Scientific). The resin was washed with 10 CVs of binding buffer and SA was eluted with 4 CVs of elution buffer (20 mM potassium phosphate, pH 2.2). The eluate was dialyzed overnight into PBS at 4 °C, concentrated using a 10 kDa MWCO centrifugal filter, and quantified by absorbance at 280 nm.
Assembly and purification of MS2-SA VLP
The assembly of MS2-SA VLPs has been previously described32,33,34,35,36. A 20 × molar excess of SA was prepared in a small glass vial at \(\ge\)30 mg/mL, and MS2 at 700 μg/mL was added dropwise under rapid stirring. The resulting mixture was run on a Superdex 200 Increase 10/300 column equilibrated in PBS to separate free SA from assembled MS2-SA particles. To quantify the amount of SA within the VLP eluate, VLP samples were mixed with Nu-PAGE lithium dodecyl sulfate (LDS) sample buffer (Invitrogen), heated at 90 °C for 30 min, and resolved on a 4–12% Bis-Tris gel (Invitrogen). The gel included SA standards of known concentrations, and band intensities were compared to determine the amount of SA incorporated into MS2-SA VLPs.
Preparation of VLP-S
The stoichiometric ratio of MS2-SA to biotinylated S was optimized by analytical SEC. Fixed amounts of biotinylated S (8 μg) were incubated with varying quantities of MS2-SA and were run on SEC. The optimal ratio was defined as the lowest amount of MS2-SA that resulted in complete incorporation of S, as indicated by the absence of unbound S in the chromatogram.
Enzyme-linked immunosorbent assay (ELISA)
Nunc MaxiSorp 96-well flat-bottom plates (Invitrogen) were coated overnight at 4 °C with 0.2 µg S protein in 100 µL PBS per well. Plates were blocked with 200 µL PBST (PBS with 0.05% Tween-20) containing 5% (w/v) bovine serum albumin (BSA) for 1 h at room temperature. 100 µL of PBST with 1% BSA containing primary antibody m336 (2 nM), or CDC2-C2 (2 nM), or S2P6 (3 nM) was added to each well. After 1 hour, wells were washed three times with PBST and incubated for 1 h with 100 µL horseradish peroxidase–conjugated anti-human IgG Fc goat antibody (MP Biomedical, cat. #674171) at a 1:5000 dilution in PBST with 1% BSA. Following three washes, wells were developed with 100 µL TMB substrate solution (Thermo Scientific) for 2.5 min, and the reaction was stopped with 100 µL of 160 mM sulfuric acid. Absorbance at 450 nm was measured using a Synergy H4 plate reader with Gen5 2.07 software (BioTek).
Sodium Dodecyl Sulfate polyacrylamide gel electrophoresis (SDS-PAGE)
Samples containing either 1 \({\rm{\mu}}\)g S or 1 \({\rm{\mu }}\)g S displayed on assembled VLP-S were prepared. Proteins were deglycosylated using PNGase F (New England Biolabs) according to the manufacturer’s denaturing reaction protocol. Following deglycosylation, samples were supplemented with 2 μL 2-mercaptoethanol and 5 μL Nu-PAGE lithium dodecyl sulfate (LDS) sample buffer (Invitrogen), then heated at 98 °C for 30 min. Proteins were separated on 4–12% Bis-Tris gels (Invitrogen) alongside a PageRuler Plus Prestained Protein Ladder (Thermo Scientific). Electrophoresis was performed in MES-SDS buffer at 4 °C for 65 min at 110 V. Gels were stained with Imperial Protein Stain (Thermo Scientific) for 15 min, destained overnight, and imaged using a ChemiDoc MP system with Image Lab 5.2.1 software (Bio-Rad).
Dynamic light scattering (DLS)
S and VLP-S proteins were diluted in 100 \({\rm{\mu }}\)L of PBS so that the final solution contained 4 μg of S. MS2-SA was diluted in 100 \({\rm{\mu }}\)L of PBS so that the solution contained 3 \({\rm{\mu }}\) g of the protein. The protein solution was added to a UVette (Eppendorf) and 13 acquisitions per sample were measured and displayed as % volume with a Zetasizer Nano (Malvern).
Analytical SEC
S and VLP-S proteins were diluted in 950 \({\rm{\mu }}\)L of PBS so that the final solution contained 8 μg of S. The solution was run on Superdex 200 Increase 10/300 Column (Cytiva) using the Unicorn 7 control system (Cytiva) and was eluted with 1 CV of PBS at 0.65 mL/minute. The absorbance at 210 nm was monitored during elution.
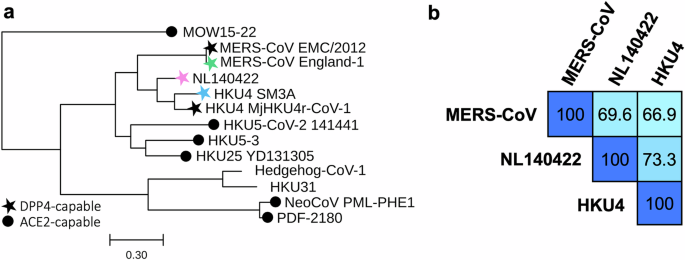
Phylogenetic tree
Spike protein amino acid sequences (Supplementary Table 2) were aligned using Clustal Omega 1.2.2. Pairwise amino acid identity percentages were calculated directly from this alignment. Phylogenetic relationships were inferred using PhyML v2012.04.12 (University of Montpellier) with a maximum parsimony starting tree and the LG amino acid substitution model. The SARS-CoV-2 spike protein (YP_009724390.1) was included as an outgroup to root the tree and subsequently removed for clarity. Trees were visualized with TreeViewer v2.2.0.
Antigenic cartography
Antigenic cartography was generated with IgG endpoint titer data using the Racmacs R package (Racmacs 1.1.35 with R 4.2.1 and RStudio 2022.07.01)35,47. The IgG endpoint titers from each serum sample against each viral antigen were used to calculate the dissimilarity value \({D}_{{ij}}\) between each serum i and antigen j according to the equation \({D}_{{ij}}=\,-{\log }_{2}{H}_{{ij}}+{\log }_{2}{H}_{i,\max }\), where \({H}_{{ij}}\) is the IgG endpoint titer of serum i against antigen j and \({H}_{i,\max }\) is the maximum titer observed for serum i. The error function for each serum-antigen pair was defined as \({E}_{{ij}}={({D}_{{ij}}-{d}_{{ij}})}^{2}\) with \({d}_{{ij}}\) denoting the Euclidean distance between serum i and antigen j on the two-dimensional map. Conjugate gradient descent optimization was applied to minimize the total error, and 5000 restarts with randomized starting positions were performed to approximate the global optimum.
Biosafety and approvals
All experiments with MERS-CoV were conducted under biosafety level 3 agriculture (BSL-3 AG) containment at the Influenza Research Institute using protocols approved by the University of Wisconsin–Madison Institutional Biosafety Committee. The facility is designed to meet or exceed the standards outlined in Biosafety in Microbiological and Biomedical Laboratories, 6th edition. All mouse studies were performed under protocol V006426 reviewed by the Institutional Animal Care and Use Committee at the University of Wisconsin-Madison.
Cell line and virus
MERS-CoV strain EMC/2012 (BEI Resources, NR-44260) was propagated in Vero E6/TMPRSS2 cells (Japanese Cancer Research Resources Bank, JCRB1819) maintained in high-glucose Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), antibiotic-antimycotic solution, and 1 mg/ml geneticin (G418; Invivogen). Vero E6/TMPRSS2 cells were tested monthly for mycoplasma contamination by PCR and consistently confirmed negative.
Animal care, immunizations, challenge studies, and antibody responses
Mice were maintained under standard conditions with ad libitum access to food, water, bedding, and enrichment. Rooms were kept at controlled temperature and humidity on a 12 h light/dark cycle. Animals were monitored at least once daily by trained personnel, and humane endpoints were applied for signs of severe infection. Group sizes were based on prior vaccine studies32; no statistical power calculations were performed. Blinding was not implemented, as all procedures were conducted under BSL-3 AG containment.
Groups of five female C57BL/6 mice (Jackson Laboratories) or hDPP4-Tg mice (colony established from 288/330 + /+ breeders kindly provided by Dr. Ralph Baric38), aged 6–8 weeks, were immunized subcutaneously under isoflurane anesthesia. Each animal received 125 μL formulations containing either 7.5 μg S displayed on VLP-S or the corresponding amount of VLP control, mixed 1:1 with an adjuvant formulation (Alhydrogel).
For immunogenicity studies, mice were bled under deep isoflurane anesthesia four weeks post-immunization. Heat-inactivated sera were analyzed by ELISA to determine antibody endpoint titers against the indicated viral S proteins. Virus neutralization assays were performed with MERS-CoV on Vero E6/TMPRSS2 cells using two-fold diluted serum samples with approximately 1000 pfu of virus. The virus and sera mixture was incubated at 37 °C for 30 min and then added to confluent Vero E6/TMPRSS2 cells that had been plated the day prior in 96-well plates. The cells were then incubated for 3 days at 37 °C. Virus neutralization titers were determined as the highest serum dilution that completely prevented CPE.
For challenge studies, hDPP4 mice were infected intranasally with 105 pfu of MERS-CoV while under isoflurane anesthesia, four weeks after immunization. At three days post-infection, mice were euthanized by isoflurane overdose, and lung and nasal turbinate tissues were collected. Viral titers were determined from clarified tissue homogenates applied to confluent Vero E6/TMPRSS2 cells overlaid with 1% methylcellulose.