
Compliance with ethical regulations
Animal care and experiments were carried out in accordance with the regulations of the applicable animal welfare acts and using protocols approved by the responsible regulatory authority (Regierung von Oberbayern, commission number 15). For human data, the collection of samples was approved by the local ethics committees of the LMU, Munich (ethical vote: 163-16, equivalent of the Institute Review Board (IRB) number for the ethics committee). Written informed consent was obtained from all individuals according to the Declaration of Helsinki.
Contact for reagent and resource sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the corresponding authors, M.K. and A.K. Plasmids generated in this study are available upon reasonable request.
Experimental model and animal details
Plasmids
All primer sequences are listed in Supplementary Table 1.
The MSCV-v2-U6-(BbsI)-Pgk-Puro-T2A-eGFP (with v2 improved scaffold) vector was generated as follows: first, the pU6-Pgk-Puro-T2A construct was PCR amplified from pKLV2-U6gRNA5(BbsI)-PGKpuro2ABFP-W (Addgene), and the eGFP construct was PCR amplified from pMSCV-Cas9-eGFP (in house); then, the PCR product was assembled into the SalI (NEB) + XhoI (NEB) digested pMSCV-neo (Takara Clontech) vector using the Gibson Assembly Master Mix (NEB). The pMSCV-v2-U6-(BbsI)-Pgk-Puro-T2A-BFP (with v2 improved scaffold) vector was generated as follows: the pU6-Pgk-Puro-T2A-BFP construct was PCR amplified with overhangs for SalI + XhoI from pKLV2-U6gRNA5(BbsI)-PGKpuro2ABFP-W, then the PCR product was digested and ligated into SalI + XhoI digested pMSCV-neo vector with Quick Ligase (NEB). The pMSCV-v2-U6-(sgNon-Targeted)-Pgk-Puro-T2A-Tdtomato (with v2 improved scaffold) vector was generated as follows: first, the pU6-Pgk-Puro-T2A construct was PCR amplified from pKLV2-U6-(sgNon-Targeted)-PGKpuro2ABFP-W, and the Tdtomato construct was PCR amplified from pAAV-CAG-tdTomato (Addgene); then, the PCR product was assembled into the SalI + XhoI digested pMSCV-neo vector using the Gibson Assembly Master Mix. The pXPR_053-hPgk-VEX vector was obtained from Addgene68. The pMSCV-v2-U6-(BbsI)-Pgk-Grx1-roGFP2 vector was generated as follows: the pLPCX cyto Grx1-roGFP2 (Addgene) and pMSCV-v2-U6-(BbsI)-Pgk-Puro-T2A-BFP vectors were digested with BglII + ClaI and the Grx1-roGFP2 and pMSCV-v2-U6-(BbsI)-Pgk constructs were ligated with Quick Ligase. The pMSCV-v2-U6-(BbsI)-Pgk-mTFP1 vector was generated as follows: the mTFP1 construct was PCR amplified from pLJM1-FIRE-pHLy (Addgene), then the PCR product was assembled into the NotI + XhoI digested pKLV2-U6-(sgRNA)-PGKpuro2ABFP-W vector using the Gibson Assembly Master Mix.
Animals
C57BL/6 mice were purchased from Charles River or Janvier and bred in the Core Facility for Animal Models of the Biomedical Center, LMU. R26-Cas9-eGFP animals were ordered from The Jackson Laboratory (024858) and backcrossed onto a C57BL/6 background several times. BiozziABH mice, F1 C57BL/6J mice crossed with BiozziABH mice, and 2D2 mice were bred in house. Animal care and experiments were carried out in accordance with the regulations of the applicable animal welfare acts and using protocols approved by the responsible regulatory authority (Regierung von Oberbayern, commission number 15). All animals had free access to food and water. Animals were kept at a room temperature of 22 °C ± 2 °C and humidity of 55% ± 10%, with a 12-h/12-h light–dark cycle (6:30–18:30). Male and female mice between 2 and 8 months old at the start of the experiment were used.
Cell lines and primary cells
All cells were incubated in a humidified incubator at 37 °C and 5% CO2 in air. All media were supplemented with 10% FBS (Bio&SELL) and 1% penicillin–streptomycin (Thermo Fisher). HEK293T cells (American Type Culture Colllection) were kept in DMEM GlutaMAX (Thermo Fisher). Primary bone marrow cells and Hoxb8FL cells were kept in RPMI GlutaMAX (Thermo Fisher). Hoxb8FL cells were additionally supplemented with 0.1% 2-mercaptoethanol (Thermo Fisher), 1 μM β-estradiol (Sigma) and supernatant from a Flt3L-producing B16 melanoma cell line, to a final concentration of 35 ng ml−1. During macrophage differentiation the RPMI medium was additionally supplemented with M-CSF (10–20 ng ml−1; PeproTech). T cells were kept in RMPI 1640 (Sigma) and additionally supplemented with 10 mM HEPES, 2 mM L-glutamine, 1% non-essential amino acids, 1 mM sodium pyruvate and 50 µM β-mercaptoethanol. For freezing cells, a 10% dimethylsulfoxide (Sigma) and 90% FBS (Bio&SELL) mixture was used, and cryovials were kept in a freezing box for several days at −80 °C before transferring to liquid nitrogen. Cell lines were tested for the absence of mycoplasma. HEK239T cells were detached by using 0.05% Trypsin-EDTA (Thermo Fisher). Macrophages were detached with Accutase solution (Sigma).
Hoxb8FL generation
hBCL2-overexpressing Hoxb8FL Cas9 (Hoxb8FL) lines were generated as follows. Bone marrow cells were harvested from femurs and tibias of 6–10-week-old animals and cultured in RPMI supplemented with recombinant mouse IL-3 (5 ng ml−1), IL-6 (20 ng ml−1) and 1% cell culture supernatant from SCF-producing B16 melanoma cells. After 2 days, the cells were spin infected with MSCV-ERHBD-Hoxb8FL-carrying retrovirus. A day after spin-infection, the cells were cultured in Hoxb8FL medium until infected cells were enriched in the culture in the presence of β-estradiol20. The medium was replaced every 2–3 days. The hBCL2 overexpression improved the survival of these cells both during in vitro differentiation experiments and in vivo.
Individual sgRNA cloning
All sgRNA sequences are listed in Supplementary Table 1.
For individual sgRNA cloning, 20-nucleotide-long sgRNAs were picked from the GPP sgRNA designer tool from the Functional Genomics Consortium of The Broad Institute, Massachusetts, USA. sgRNA sequence and reverse complemented sequence were ordered as two separate oligonucleotides from Metabion with overhangs on the 5′ side of CAAC for forward and AAAC for reverse. The nucleotide ‘G’ was added as a first nucleotide to increase the efficiency of the gRNA expression by the hU6 promoter79. Complementary oligonucleotides with overhangs were phosphorylated and annealed in the presence of 10x T4 Ligation Buffer (NEB) and T4 PNK (NEB) by increasing the temperature to 95 °C and ramping down to 25 °C at 5 °C min−1. Annealed oligonucleotides were ligated into Bpil- or BsmbI-digested (Thermo Fisher) gRNA cargo plasmid by Quick Ligase (NEB) for 6 min at room temperature. Ligated plasmids were then transformed into Stellar competent cells (Takara Clontech) with heat shock at 42 °C for 55 s. Bacterial plates were incubated overnight, and single clones were picked and prepped with the Qiagen plasmid miniprep kit. The correct ligation product was confirmed with Sanger sequencing (Sequencing service, LMU Biozentrum) using the hU6 primer.
Oligonucleotide pool library cloning
sgRNAs were picked from the GPP sgRNA designer tool from Functional Genomics Consortium of The Broad Institute. All sgRNA sequences that were selected, and the library cloning primers, as well as protocols, are listed in Supplementary Table 1.
sgRNAs for the mouse Cytokine Receptor library were ordered from Integrated DNA Technologies as custom oPools (50 pmol per oligonucleotide) as a 79-mer with a sequence of 5’-GCAGATGGCTCTTTGTCCTAGACATCGAAGACAACACCGN20GTTTTAGTCTTCTCGTCGCC-3’, with N20 indicating the sgRNA sequence. Each gene was targeted with three different sgRNAs (Stat6 with 4 sgRNAs), and 15 non-targeted (from here onward, control) control gRNAs were also included (343 oligonucleotides in total). The oligonucleotide pool was dissolved in Qiagen TE buffer to get a 100 μM stock concentration, and then the single-stranded oligonucleotides (100 ng) were PCR amplified for two cycles with Q5 High-Fidelity DNA Polymerase (NEB) using Oligo_Amp_F and Oligo_Amp_R primers to generate double-stranded DNAs. The PCR products were purified with the Nucleotide Removal Kit (Qiagen). Amplified double-stranded DNAs were digested with FastDigest BpiI (BbsI, Thermo Fisher) for 2 h at 37 °C in a total of two reactions, and then purified with the Nucleotide Removal Kit (Qiagen). Ligation was performed with a T4 DNA Ligase (NEB) using a 3 ng insert and 40 ng BpiI-digested MSCV-v2-U6-(BbsI)-Pgk-Puro-T2A-eGFP for 16 h at 16 °C per reaction in a total of two reactions. The ligated product was cleaned with a PCR Purification Kit (Qiagen), and the concentration was measured with Qubit 4 (Thermo Fisher). Ten nanograms of the ligated product was transformed into 50 μl of NEB Stable Competent cells in a total of eight reactions and incubated at 30 °C overnight. A library representation above 100× was confirmed by plating transformed competent cells in serial dilutions. The plasmid DNA was prepared with an Endofree Plasmid Maxi Kit (Qiagen).
Gene editing of Hoxb8FL cells
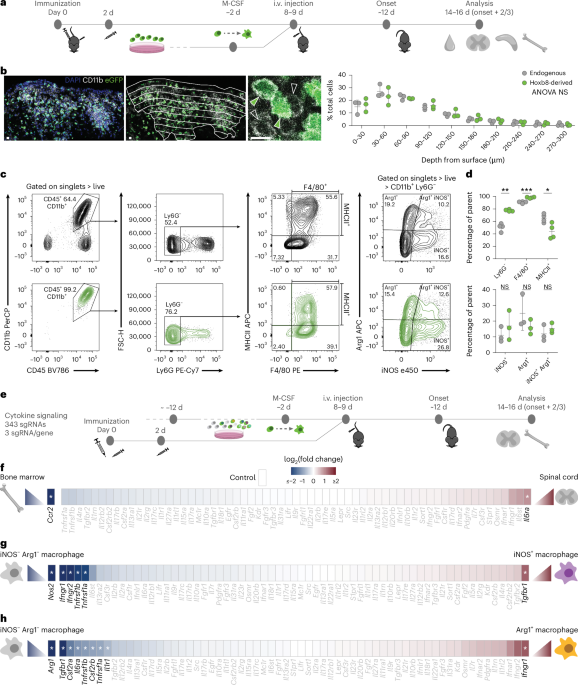
For viral transduction of Hoxb8FL cells, HEK293T cells were plated into a six-well plate 18–24 h before transfection. For the transfection per well of six-well plate, 1.5 µg pMSCV retroviral plasmid and 1.5 µg pCL-Eco packaging vector were added in 400 µl of RPMI medium without serum and antibiotics. Then 7.5 μl of the transfection reagent TransIT-LT1 were added into the mix, vortexed, and then incubated at room temperature for 30 min. After 30 min, the solution was added dropwise to HEK293T cells. After 4 h, the medium was replaced with Hoxb8FL medium. Retrovirus-containing supernatant was collected at 48–72 h after transfection. Hoxb8FL cells were spin infected with freshly harvested virus at 1,200g for 30 min at room temperature. For individual KO cell line generation, individual sgRNA viruses were produced and transduced. For the CRISPR screen experiments, the library oligonucleotide pool was used to generate a virus mix containing all the library sgRNAs, which was then transduced at a maximum multiplicity of infection of 0.3 or below (< 30% transduction efficiency) to prevent multiple integrations of sgRNAs into a single cell, and enough cell numbers were always kept ensuring a minimum 1,000× coverage (1,000 cells having the same sgRNA). For the Perturb-seq experiment, viruses for six individual non-targeted sgRNAs and two different sgRNAs per gene targeted were produced individually, and single KO or control cell lines were generated in parallel and mixed in equal proportions before expansion before i.v. injection. On the next day, puromycin (Thermo Fisher) was added at a final concentration of 5 µg ml−1 to select for transduced cells. After 4 days of puromycin selection, cells expressing fluorescent markers (for example, eGFP) were sorted to purity to ensure high expression of the fluorescent marker by using a FACS Aria III (BD) or FACS Fusion (BD) at the Flow Cytometry Core Facility of the Biomedical Center, LMU. The combination of positive selection of the infected cells by puromycin with sorting of the pure fluorescent population ensured all the Hoxb8FL transferred cells in the in vivo experiments were gene edited.
Tide assay
All single KO lines used in validation experiments were assessed for KO efficiency before the experiment with the TIDE assay. This assay works as follows: to assess the extent of genetic editing at DNA level for single sgRNAs, genomic DNA from sgRNA transduced (see above) Hoxb8FL cells was isolated with the DNeasy Blood and Tissue Kit (Qiagen), and the DNA region targeted by the sgRNA was amplified with Q5 High-Fidelity DNA Polymerase (NEB) and specific PCR primers for each different sgRNA. All TIDE primers are listed in Supplementary Table 1. The samples from KO and control cells were submitted to Sanger Sequencing with either the forward or the reverse PCR primer (Sequencing service, LMU Biozentrum). The Cas9-dependent generation of insertion–deletion repair errors and the subsequent KO efficiency (frame shifts that are not a multiple of 3 are considered to generate a protein KO) were assessed by the ICE v2 software tool80.
Intravenous transfer of Hoxb8FL cells
In vitro-expanded Hoxb8FL cells were washed twice with PBS to remove β-estradiol and cultured in RPMI medium (with 10 ng ml−1 M-CSF) for 2 days to initiate myeloid differentiation. On day 8–9 after active EAE induction, or at disease onset in TH17 adoptive transfer EAE (onset of clinical symptoms or >1 g of weight loss, or else the Hoxb8FL transfer stopped disease development), cells were washed twice with PBS and 10–15 × 106 cells in 200 μl of PBS were injected i.v. into the tail vein of the mice.
For the intravital imaging experiments, Hoxb8FL cells were labeled before transfer with CellTrace Far Red (Thermo Fisher Scientific, C34564), CellTrace Yellow (Thermo Fisher Scientific, C34567) or CellTrace Violet (Thermo Fisher Scientific, C34571) for in vivo detection. After reconstituting the CellTrace according to the manufacturer’s instructions, cells were collected and the residual medium was washed once with PBS. Then, the cells were incubated 30 min at 37 °C in the dark in a 1:500 dilution of CellTrace solution in PBS at a concentration of 5 × 106 cells per ml. Staining was stopped by adding five times the volume of cell medium and incubating for 5 min at 37 °C in the dark, followed by two PBS washes before injection.
Flow cytometry
Tissue isolation from EAE animals
Animals with EAE clinical signs were euthanized by isoflurane overdose. When CSF was collected, it was collected immediately after euthanasia from the cisterna magna. Briefly, the animal was head-fixed in a stereotactic frame, and the cisterna magna was exposed by keeping the head bent down at a 45° angle and removing skin and muscle from the neck. Then, a pulled glass pipette was used to carefully penetrate the cisterna magna and suction the CSF. Between 10 μl and 25 μl of CSF was collected per animal and immediately resuspended on FACS buffer (0.5% BSA, 1 mM EDTA, 25 mM HEPES on calcium and magnesium-free PBS) on ice. Blood samples were collected immediately after euthanasia or after CSF extraction by cardiac puncture, and the animal’s body was perfused with PBS-heparin before collecting the spleen, inguinal lymph nodes, bone marrow (femur and vertebra), spinal cord and brain cortex through microdissection. Cells from bone marrow were collected by flushing with ice-cold PBS, whereas cells from vertebrae were isolated after crushing with a pestle and mortar. Spleen, lymph nodes, spinal cord and brain tissues were homogenized with a glass dounce homogenizer (Wheaton) with a loose pestle, transferred into a PBS solution containing collagenase D (0.8 mg ml−1; Roche) and DNase I (10 ng ml−1; Roche) and incubated for 20 min at 37 °C with shaking (1,000 rpm) for dissociation of cells. For scRNA experiments, cells were incubated in digestion buffer for only 5 min to minimize changes to the transcriptome. Cells from spleen, lymph nodes (after digestion) and bone marrow were treated with ACK Lysing Buffer (Thermo Fisher) for 5 min, whereas cells from blood were incubated for 20 min on ice for red blood cell lysis. All cell suspensions were passed through a 100-μm-pore-diameter cell strainer (Corning). Cells from spinal cord and brain were isolated with Percoll (Sigma) gradient to remove the myelin. Cells were resuspended with 1 ml of 100% FBS and 9 ml of 33% Percoll (in PBS), and 1 ml of 10% FBS (in PBS) was added on the top slowly to form a layer. Samples were centrifuged without brakes at 800g for 15–30 min at 4 °C. The myelin layer was carefully sucked with a vacuum, and pelleted cells were washed with PBS to remove the Percoll solution.
CRISPR library experiments
For the cytokine library experiment, around ten animals were combined for one replicate to ensure sufficient cells (as, depending on the disease score, cell numbers found in the spinal cord vary). Before starting flow cytometry labeling, monocytes/macrophages from the spleen and bone marrow were enriched with MACS using anti-CD11b microbeads (Milteny) to reduce sorting time. All cells were blocked with TruStain fcX (anti-mouse CD16/CD32, BioLegend) and stained with Live/Dead Near-IR (Thermo Fisher) to exclude dead cells during blocking for 30 min at 4 °C. After washing with PBS, cells were incubated with antibodies recognizing extracellular markers for 30 min at 4 °C, with anti-CD11b-PerCP antibody to include monocytes/macrophages and anti-Ly6G-BV786 antibody to exclude granulocytes. Cells from the spinal cord were then fixed and permeabilized using Cytofix/Cytoperm kit (BD Biosciences) for 15 min at 4 °C, before intracellular labeling. Cells were incubated with anti-Arg1-APC and anti-iNOS-Pacific Blue antibodies for 30 min at 4 °C in Perm/Wash buffer solution. Hoxb8FL-derived cells were identified by their eGFP fluorophore expression. Populations of interest were isolated using a FACS Aria III (BD) or FACS Fusion (BD) at the Flow Cytometry Core Facility of the Biomedical Center. Enough cells were sorted for each population to ensure a library coverage of a minimum of 100× (100 cells containing each sgRNA). Genomic DNA from sorted cells was isolated by using the QIAamp DNA Micro Kit. Cells were lysed in the presence of Proteinase K at 56 °C overnight (modified from the manufacturer’s protocol) for more efficient de-crosslinking of fixed DNA before isolation of genomic DNA.
In vitro polarization
For in vitro macrophage polarization experiments, bone marrow-derived cells were cultured in RPMI medium with 10 ng ml−1 M-CSF for 5–7 days to allow differentiation. Differentiated cells were then re-seeded and the respective recombinant mouse cytokines were added at a concentration of 10–20 ng ml−1 as reported in the literature81 for 48 h before FACS: IL-4 (R&D Systems), TGFβ (BioLegend), GM-CSF (PeproTech), IFNγ (R&D Systems) or TNF (PeproTech) were used. Cells were detached with Accutase followed by two washing steps with PBS. Cells were fixed and permeabilized using the Cytofix/Cytoperm kit for 15 min at 4 °C before incubation with anti-Arg1 and anti-iNOS antibodies for 30 min at 4 °C in Perm/Wash buffer solution. Cells were analyzed with a Fortessa (BD) or a Cytoflex S (Beckman Coulter).
In vivo Hoxb8FL-derived cell characterization
For characterization experiments of Hoxb8FL-derived cells, nonspecific binding was blocked with TruStain fcX (anti-mouse CD16/32, BioLegend) and cells were stained with Live/Dead reagent (Thermo Fisher) to exclude dead cells, for 30 min at 4 °C. After washing with PBS, cells were incubated with antibodies recognizing extracellular markers for 30 min at 4 °C: anti-CD11b, anti-CD45, anti-Ly6G, anti-Ly6C, anti-MHC class II and anti-F4/80 were used. Cells then were incubated with anti-Arg1 and anti-iNOS antibodies for 30 min at 4 °C in Perm/Wash buffer solution. All antibodies—unless otherwise stated—were used at 1:100 dilution. Hoxb8FL-derived cells were identified by eGFP fluorophore expression, and eGFP-negative cells were assigned as the endogenous population. Cells then were analyzed using a FACS Aria III (BD) or FACS Fusion (BD) at the Flow Cytometry Core Facility of the Biomedical Center. Neutrophils were excluded from all analyses by expression of Ly6G.
For single KO validation experiments, before the transfer of Hoxb8FL cells into animals, Hoxb8FL cells expressing tdTomato and control sgRNA were mixed at a 1:1:1 ratio with Hoxb8FL cells expressing either BFP or eGFP and the gene targeting sgRNA to minimize the inter-animal experimental variation.
scRNA-seq experiments
For the 10x scRNA-seq experiments, before euthanasia, 3 μg per mouse of anti-CD45 antibody was injected i.v. in the tail vein to permit exclusion of cells derived from blood. Cells from 7 mice were pooled from a single EAE round for the WT EAE experiment; cells from 6 (round 1) and 12 (round 2) mice were pooled from two independent Hoxb8FL cell generation and EAE rounds for the Hoxb8FL-derived cell experiment; for the chimera experiment, three mice each with control, Ifngr1-KO, Tnfrsf1a-KO and Csfr2ra-KO BM chimera, and two mice with Tgfbr1-KO BM chimera were pooled; for CSF sequencing, 11 (round 1) and 12 mice (round 2) were pooled; for cEAE, 3 healthy, 3 d3 cEAE and 3 d14 cEAE mice were pooled. All cells were incubated with TruStain fcX (anti-mouse CD16/32, BioLegend) and Live/Dead Near-IR (Thermo Fisher) to exclude dead cells for 30 min at 4 °C. After washing with PBS, cells were incubated with anti-CD11b-PerCP antibody to include monocytes/macrophages and anti-Ly6G-BV786 antibody to exclude granulocytes, for 30 min at 4 °C. For the cEAE experiment, cells were additionally incubated with CD45-PE antibody to include all immune cells. Hoxb8FL-derived cells were identified by eGFP fluorophore expression, and eGFP-negative cells were assigned as the endogenous population. Chimeric cells were identified by Vex fluorophore expression. Populations of interest (WT, Hoxb8FL and chimeric EAE: CD11b+Ly6G−; CSF and cEAE CD45+Ly6G−) were sorted for purity using a FACS Aria III (BD) or FACS Fusion (BD) at the Flow Cytometry Core Facility of the Biomedical Center. Sorted cells were immediately prepared for 10x single-cell experiments according to the 10x protocol. The gating strategy to sort for Ly6G− cells resulted in minimal numbers of neutrophils in the spinal EAE scRNA-seq experiments: 310 cells, 2% of total, in the WT dataset, 31 cells, 0.28% of total, in the Hoxb8FL-transfer dataset and 174 cells, 0.53% of total, in the bone-marrow chimera dataset.
Statistical analysis and software
No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those reported in previous publications8,82,83. For statistical analyses and plotting, GraphPad Prism versions 7 and 9 (GraphPad Software) and R84 (version 4.0.0+) were used. For next-generation sequencing sample demultiplexing, Je-Demultiplex85 was used in Galaxy86. For analysis of CRISPR screens, cutadapt87, trimmomatic88, the MAGeCK software89, Galaxy86 and R84 were used. For analysis of bulk Galaxy86, RNA STAR90 (version 2.7.2b in Galaxy), HTSeq-count91 (version 1.0.0 in galaxy), DESeq2 (ref. 92; version 2.11.40.7 + galaxy1) and R84 were used. For image analysis, Fiji/ImageJ93 and Imaris (Oxford Instruments) were used. For scRNA-seq data, Seurat94,95,96,97,98 (versions 4+) and R84 were used. Figures were made using the aforementioned software and Adobe Illustrator 2025. Data are represented as the mean ± standard deviation unless otherwise stated; box plots show the median and quartiles 1 and 3 for the box limits, and whiskers extend to 1.5 times the interquartile range. Sample sizes are reported in the figure legends. All replicates are biological unless otherwise stated, measurements were not repeated, and all tests are two tailed unless otherwise stated. The choice to do a one-tail or two-tailed test depended on whether there were prior data suggesting the expected effect would be in a specific direction. Asterisks indicate P value > 0.05 (NS), *P value < 0.05, **P value < 0.01, ***P value < 0.001 and ****P value < 0.0001, unless otherwise specified. Samples were tested for a normal distribution using the Shapiro–Wilk normality test, and the statistical test used were chosen so the data met the assumptions of the test. Animals were randomly assigned to experiments or, when not possible, the conditions were appropriately blocked such that there were animals of the same condition in all cages. Due to the experimental design and animal laws, no blinding was possible during FACS experiments and Hoxb8FL characterization histology. The analysis of all histological data was done blinded. scRNA-seq exclusion criteria for low quality control are detailed in the Supplementary Methods. No other data were excluded. For statistical testing of the cell distribution in the lesion (Fig. 1b), an ordinary two-way ANOVA with the two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli for multiple comparisons was used. For the Hoxb8FL transfer experiments (Figs. 1, 2, 5 and 6 and Extended Data Figs. 1, 2 and 7), multiple paired t-tests or Wilcoxon tests (when the sample was not normally distributed) with the two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli for multiple comparisons with FDR (q) of 5% when needed were used. For comparison of EAE disease course in WT and Hoxb8FL transferred animals (Extended Data Fig. 1o), the Kolmogorov–Smirnov test was used. For the intravital imaging experiments (Figs. 4 and 5), for the proportion of cells in each ratio bin, an ordinary two-way ANOVA with a single pooled variance, for the interaction of genotype and bin was run; a Kolmogorov–Smirnov test was run for cumulative distributions, and a one-tailed unpaired (Fig. 4) or two-tailed paired (Fig. 5) t-test for the proportion of cells above Q3 of the ratio. For the histological quantifications, paired t-test or Wilcoxon tests were run. For the cholesterol efflux assay, unpaired t-tests were run. For the pHrodo-myelin phagocytosis assay, an ordinary two-way ANOVA with a single pooled variance for the genotype differences was run. For the single KO phenotype differences in polarization between Hoxb8FL and chimera, unpaired t-tests or Wilcoxon tests were run. For the KO/control distribution phenotype (Fig. 6), paired t-tests or Wilcoxon tests comparing KO and control were used, but data are plotted as the KO/control ratio for visualization purposes only. For the motility analysis, paired t-tests were run. For CRISPR screen results (Figs. 1 and 2 and Extended Data Figs. 2 and 3), the MAGeCK software was used as described above. Asterisks indicate P value and FDR < 0.05 and absolute log2(fold change) > 3 times the standard deviation of the noise distribution, as previously described. For comparison of in vitro bone marrow-derived macrophage polarization conditions (Extended Data Fig. 4), an ordinary one-way ANOVA with the two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli for multiple comparisons was used. Bulk RNA-seq differentially expressed genes (Extended Data Fig. 4) were defined as having an adjusted P value < 0.05 and log2(fold change) > 3 times the standard deviation of all log2(fold change) in the comparison. For individual gene KO-versus-control phenotypes in the scRNA-seq experiments (Extended Data Figs. 5, 6 and 7), the FindMarkers function of the Seurat package (version 5) was used with default parameters other than logfc.threshold = 0 and min.pct = 0.1, and differentially expressed genes were defined as adjusted P value < 0.05 and absolute log2(fold change) > 3 times the standard deviation of the log2(fold change) distribution of the cluster and as described above for Fig. 7. To evaluate the significance of the enrichment of the signatures in the KOs versus the controls (Figs. 4 and 5), the GSEA preranked function of the GSEA software99,100 (version 4.2.3) was used, with a custom-made database of the signatures, as described elsewhere. A signature was considered significantly enriched when the NOM P value < 0.05 or the FDR q value < 0.25 and absolute NES > 1.5. For unbiased pathway analysis (Fig. 6), the pathways depicted in the figure were filtered based on the output parameters of gprofiler101 as described above. For statistical analysis of human samples (Fig. 8), the Shapiro normality test was used to evaluate the normal distribution of the sample, and after, according to the results, a one-way ANOVA was performed for each cytokine signature for newly generated CSF samples followed by t-tests or Wilcoxon tests comparing disease conditions against controls. For datasets from Schafflick et al.32 and Piehl et al.33, t-tests or Wilcoxon tests comparing disease conditions against controls were used. For samples from Macnair et al.35 and Mathys et al.36, the Shapiro normality test was used to evaluate the normal distribution of the sample, and after, according to the results, a one-way ANOVA or Kruskal–Wallis test per cytokine-induced signature per dataset was ran, followed by t-tests or Wilcoxon tests comparing all disease/lesion conditions to control (gray matter or healthy, respectively) and all clusters to the homeostatic cluster.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.