
Study participants
Patients with FD (Rome IV criteria) were recruited from the University Hospitals Leuven (UZ Leuven, Belgium) outpatient clinic between March 2021 and January 2024. Patients between 18 and 64 years old qualified for inclusion when FD symptoms were the predominant gastrointestinal (GI) complaints, no major inflammatory (including gastritis), metabolic or psychiatric condition was present and when Helicobacter pylori status was negative. Patients did not report allergic or atopic conditions, had no history of gastrointestinal surgery other than appendectomy, and did not take any anti-inflammatory or anti-allergy drugs ( < 2 weeks before the study); immunosuppressants, antibiotics or acid-suppressive drugs including PPI ( < 3 months before the study); or prokinetics and antacids ( < 2 weeks before the study, unless if ≤3 times per week). Women were not pregnant or lactating. Simultaneously, healthy volunteers were recruited via advertisement as controls. Similar exclusion criteria applied to healthy controls (HC), with the absence of any recurring GI symptoms in HC. All participants signed written informed consent prior to inclusion. This study was approved by the UZ Leuven Ethics Committee (S64807 and S64847), registered at ClinicalTrials.gov (NCT04713969, 13/01/2021), and conducted in accordance with Good Clinical Practice regulations and the Declaration of Helsinki.
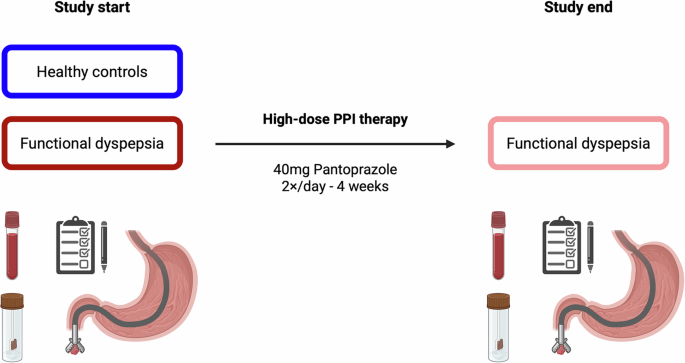
Study design and sample collection
During a baseline study visit FD patients and HC underwent a gastroduodenoscopy by an experienced endoscopist (TV) to collect duodenal (D2) biopsies and exclude the presence of endoscopic abnormalities. FD patients started a 4-week treatment with high-dose proton pump inhibitors (40 mg pantoprazole 2×/day for 4 weeks), followed by a second study visit including a gastroduodenoscopy (Fig. 1, Supplementary Fig. 1). Biopsies were collected in 10% formalin for histological analyses or in complete RPMI (cRPMI: RPMI-1640 (Gibco) with 10% fetal calf serum (Gibco), 100 U/mL penicillin and 100 µg/mL streptomycin (Lonza)) for short-term incubation and lamina propria leukocyte (LPL) isolation.
During study visits, blood was drawn by qualified staff (two EDTA Vacutainer tubes, BD). Within two days of the study visits, participants collected a fecal sample at home using previously described collection procedures28, which was immediately frozen and transported to the lab on ice packs. Participants completed online questionnaires to assess GI-specific (PAGI-SYM)29 and extraintestinal somatic (PHQ-12)30 symptoms, as well as general well-being (RAND-36 Health Survey)31, sleep quality (PSQI)32 and GI-related quality of life (PAGI-QoL)33. Patients with FD recorded their symptoms on a daily basis using the Leuven Postprandial Distress Scale (LPDS)34, starting from at least one week before the study (baseline recording) until the end of the study, reported as weekly averages. Concomitant irritable bowel syndrome (IBS) was diagnosed according to the Rome IV criteria, while the ReQuest questionnaire assessed overlapping reflux symptoms, although FD was the predominant disorder in all patients.
Peripheral blood leukocyte isolation
Peripheral blood mononuclear cells (PBMCs) and polynuclear cells (PBPCs) were isolated from fresh peripheral blood and frozen to enable batch analysis as previously described35. In brief, fresh peripheral blood was diluted in PBS (without Ca2+ and Mg2+, Gibco) supplemented with 2 mM EDTA (Invitrogen) and layered over Ficoll (Lympholyte-H Cell Separation Media, Cedarlane) before centrifugation (20 min, 800 × g, no deceleration). The resulting buffy coat was isolated by careful aspiration and washed twice with PBS-EDTA before cell counting. Mononuclear cells were resuspended in fetal calf serum (FCS, Gibco) and subsequently diluted with freezing medium (final concentration: 10% dimethyl sulfoxide (DMSO, Sigma) in FCS) under gentle swirling to a density of 4 × 106 cells/mL. Cells were frozen to −80 °C at a rate of −1 °C/min (CoolCell, Corning), before transfer to liquid nitrogen. After Buffy coat separation for PBMC isolation, the supernatant and Ficoll layers were removed by gentle aspiration to isolate circulating polynuclear cells in a subset of participants (22 HC, 17 FD before and after high-dose PPI). The remaining red blood cell fraction was incubated twice for 10 min with freshly prepared lysis buffer (in mM: 150 NH4Cl, 10 KHCO3, 1 EDTA; pH 7.4). Peripheral blood polynuclear cells (PBPCs) were washed with PBS, resuspended (4 × 106 cells/mL, 10% DMSO in FCS), and frozen as outlined for PBMCs, before storage in liquid nitrogen until analysis.
Duodenal lamina propria leukocyte isolation
Isolation of lamina propria leukocytes (LPLs) was performed based on a previously described protocol36. Four to six duodenal biopsies collected in cRPMI, were processed within 30 min after collection. Mucus and epithelium were washed by incubating biopsies twice in cRPMI containing 10 mM EDTA and 20 mM HEPES for 10 min at 37°C with magnetic stirring. Biopsies were then mechanically disrupted with fine scissors and enzymatically digested in Hank’s Balanced Salt Solution (HBSS with Ca2+ and Mg2+, Gibco) containing 20 mM HEPES, 0.2 U/mL collagenase IV (Worthington) and 10 µg/mL DNase I (Roche) at 37 °C with magnetic stirring for 30 to 45 min. The resulting single cell suspension was filtered (70 µm cell strainer) and washed in PBS + 0.5% BSA buffer before staining.
Flow cytometry
PBMC suspensions were briefly defrosted in a 37 °C water bath for flow cytometric staining, followed by suspension in thawing medium (20% FCS in RPMI-1640 (Lonza)) and centrifugation at 4 °C. Cells were resuspended in thawing medium containing DNase (100 µL/mL, Sigma) and incubated for 10 min at 37 °C. PBMCs were stained with Zombie NIR Fixable Viability dye (BioLegend), followed by staining for surface markers (Supplementary Table 7) to identify relevant mononuclear cell populations (Supplementary Fig. 3). Samples were acquired on a Cytek Aurora spectral flow cytometer coupled to SpectroFlo software (Cytek Biosciences) and analyzed in FlowJo software (BD).
PBPC suspensions were briefly defrosted in a 37 °C water bath for flow cytometric staining, followed by suspension in cRPMI containing 20 mM HEPES (Gibco) and 1% sodium pyruvate (Gibco). Cells were pelleted and washed with 0.5% bovine serum albumin (BSA, Sigma) in PBS. PBPCs were stained with Fixable Viability Dye eFluor 780 (FVD, eBioscience) for 25 min at room temperature and washed with 0.5% bovine serum albumin (BSA, Sigma) in PBS. Fc receptor blockade with heat-inactivated human plasma for 10 min at 4 °C was followed by PBS-BSA wash and staining with surface marker antibodies (Supplementary Table 8) for 30 min at 4 °C, enabling identification and phenotyping of peripheral eosinophils and neutrophils (Supplementary Fig. 4). After PBS-BSA wash, cells were fixed in 1% PBS buffered formaldehyde for 15 min at room temperature and washed in PBS-BSA containing 2 mM EDTA. Cell suspensions were kept at 4 °C until acquisition on an LSR Fortessa flow cytometer (BD) with FACSDiva software (BD) as described previously36, and analyzed in FlowJo (BD).
Fresh LPLs were stained for flow cytometry to characterize selected innate and adaptive immune populations using two different antibody mixtures (Supplementary Table 9), focusing on eosinophils, mast cells and lymphocyte subsets (Supplementary Figs. 5 and 6). The adaptive immune cell antibody panel was modified from a previously optimized panel37. LPL were incubated with FVD780 (innate) or FVD450 (adaptive, both eBioscience) for 25 min. Subsequent staining and flow cytometry procedures were identical to PBPC experiments.
Histological eosinophil and mast cell quantification
Formalin-fixed and paraffin-embedded duodenal biopsies were sectioned at 5 µm thickness at the University Hospitals Leuven Pathology Unit and subsequently stained with hematoxylin and eosin (H&E) or cKit (CD117; polyclonal rabbit anti-human antibody, 1:500 dilution, Agilent) for eosinophil and mast cell quantification, respectively. Stained slides were digitalized with an Aperio CS2 (Leica Biosystems) or Axio Scan 7 (Zeiss) scanner. Cell quantification was performed on whole-slide images using the Leuven Intestinal Counting Protocol12.
Eosinophil and mast cell mediator release
Fresh duodenal biopsies were transferred to pre-warmed cRPMI containing 20 mM HEPES (Gibco), and subsequently incubated for 24 h at 37 °C. Upon collection, biopsy supernatant was snap-frozen in liquid nitrogen and stored at −80 °C until analysis. Eosinophil and mast cell mediators were measured in biopsy supernatants using commercial eosinophil-derived neurotoxin (EDN, Diagnostics Development) and tryptase (TPSB2, ThermoFisher Scientific) ELISA kits, respectively. Mediator concentrations were normalized to biopsy weight.
Fecal EDN was measured in frozen samples according to a previously published protocol38 with following modifications. Fecal samples were thawed on ice and weighed, before 1:10 dilution in fresh ice-cold fecal extraction buffer (20% glycerol (MP Biomedicals), 1% BSA (Sigma), 0.2% cetrimonium bromide (CTAB, Sigma), 0.05% Tween 20 (Merck) and 10 mM EDTA (Millipore) in PBS (Gibco)). One 5-mm stainless steel bead (Qiagen) was added per sample for homogenization by vortexing. Next, homogenates were incubated for 30 min on ice, before additional dilution (1:100 final dilution) in fresh ice-cold fecal extraction buffer. Homogenates were centrifuged at 20,800 × g for 30 min (4 °C), before transferring the supernatant for storage at -80°C until analysis. EDN was measured in fecal extracts using ELISA (Diagnostics Development) and normalized to the semi-dry weight of fecal samples.
Isolation and culturing of murine dorsal root ganglia
Murine dorsal root ganglion (DRG) neurons were isolated as described before16, with minor modifications. Thoracic DRGs (T5-T13) from male Balb/c wild-type mice (8–12 weeks old) were bilaterally excised from the vertebral column in cold Hank’s Balanced Salt Solution (HBSS with Ca2+ and Mg2+, Gibco) under a stereomicroscope using a fine forceps. DRGs were collected in ice-cold basal medium (BM; 10% FCS in Neurobasal-A medium (Gibco); pH 7.3), containing 1 × antibiotic-antimycotic cocktail (Gibco). After collection of all DRGs, the medium was replaced with BM containing 0.8 mg/mL collagenase I (Gibco) and 1 mg/mL dispase II (Gibco), and incubated for 1 h at 37 °C while gently shaking (150 rpm). Next, DRGs were washed twice with BM and once with complete medium (CM; Neurobasal-A medium containing 2% B27 supplement (Gibco), 2 mM Glutamax (Gibco), 1 × antibiotic-antimycotic, 10 ng/mL recombinant human neurotrophin-4 (NT-4, Preprotech), and 2 ng/mL recombinant human glial cell line-derived neurotrophic factor (GDNF, Invitrogen); pH 7.3), before replacing the medium with 9% bovine serum albumin (BSA) in CM. Individual neurons were released from DRGs by gentle trituration through subsequent 22 G, 26 G and 27 G needles. The neuronal suspension was layered over 16% BSA in PBS, and debris was removed by density centrifugation (6 min, 500 × g). DRG neurons obtained from two animals were pooled in CM to increase cell yield and minimize variability, while results of stimulation experiments were obtained from two to three technical replicates. Cells were seeded on poly-D-lysine/laminin (both Sigma) coated 18-mm coverslips and incubated for 18–24 h at 37 °C (95% O2, 5% CO2) until imaging. Animal experiments were conducted in accordance with European Community Council guidelines and approved by the KU Leuven Ethics Committee for Animal Experiments (P126/2023).
Ca2+ imaging of murine dorsal root ganglia
Primary DRG neuronal cultures were loaded with 1 µM Fluo-4 AM in HEPES-buffered Krebs solution (in mM: 148 NaCl, 5 KCl, 10 HEPES, 1 MgCl2, 2 CaCl2, and 10 glucose; pH 7.4) at room temperature for 20 min while gently shaking (100 rpm), protected from light. The loading solution was replaced by HEPES-Krebs and preparations were washed for at least 10 min. Ca2+ imaging was performed using an Inverted Zeiss Axiovert 200 M microscope with a 20 × lens, coupled to a monochromator (Poly IV, TILL Photonics) and cooled CCD camera (Imago QE, Till Photonics). After baseline recording, Ca2+ transients were measured in response to human duodenal biopsy supernatants (10 s, 1:5 diluted in HEPES-Krebs), and high-K+ solution (10 s, HEPES-Krebs solution with iso-osmotic substitution of NaCl for KCl to 75 mM K+). TRPV1+ neurons were identified by application of the TRPV1 agonist capsaicin (10 s, 1 µM)16.
Image processing and analysis
Fluorescence recordings of Ca2+ imaging experiments were analyzed using custom written macros in Igor Pro software (v8, Wavemetrics). Fluo-4-labeled neuronal cell bodies were manually identified and annotated by regions of interest (ROI), in which Ca2+ transients were analyzed after correcting for background fluorescence, by normalizing fluorescence intensity to the baseline value (F/F0). Viable neurons were identified by a sharp Ca2+ transient in response to high-K+ perfusion39. Transient Ca2+ peaks in response to stimuli were considered meaningful if the peak amplitude exceeded a 10% increase compared to baseline (ΔF/F0 > 1.1).
Statistical analysis
Statistical analyses were performed in SAS Studio software (v3.8) and GraphPad Prism software (v10.2.3), while GraphPad Prism was used to create graphs. Univariate comparisons between FD and HC were performed via two-sided (un)paired t tests or non-parametric alternatives for continuous variables, and Fisher’s exact tests for categorical variables. Variables that did not follow Gaussian distribution were Box-Cox transformed to meet the assumption of normally distributed residuals. The effect of treatment was analyzed by linear mixed models with PPI treatment as within-subject factor. Time was added as within-subject factor in linear mixed models for repeated measurements (LPDS), with specification of an autoregressive correlation structure, and stepdown Bonferroni correction for pairwise post-hoc tests compared to baseline. Correlation analyses (Spearman’s correlation coefficients ρ) were performed in R software (v4.4.2). A P < 0.05 was considered statistically significant.