
Defective in vivo response to eTLRs in Munc13-4 deficiency
We previously showed that Munc13-4 binding to STX7 regulates the eTLR response to CpG16. Munc13-4 polymorphism is associated with MAS, sJIA and systemic inflammation, while chronic TLR9 stimulation in mice develops features characteristic of MAS, including elevated serum levels of IFNγ, interleukin 6 (IL-6) and hepatic inflammation10. Here, to study whether TLR9 activation is regulated by Munc13-4 in vivo, we treated Munc13-4-null (Munc13-4Jinx/Jinx) mice with a single CpG insult and analyzed the neutrophilic and cytokine-mediated inflammation. In these studies, cytokine production was analyzed 6 h after CpG insult, which is a well-established time point of maximal cytokine production in mice challenged with CpG31. TLR9 stimulation of Munc13-4Jinx/Jinx mice results in reduced cytokine production, including IL-6, IFNγ and IFNα, compared to wild-type mice (Fig. 1a–c). IL-12, although reduced, did not reach significance (Fig. 1d). This differs from that observed in TLR4-initiated systemic inflammation in Munc13-4-null mice32. Thus, the production of proinflammatory cytokines and tissue infiltration by myeloid cells were normal (similar to wild type) in Munc13-4-deficient mice when challenged, in vivo, with the TLR4 ligand lipopolysaccharide (LPS)32. Despite normal neutrophil counts (Fig. 1e), neutrophil activation measured as the plasma level of the azurophilic granule cargo myeloperoxidase (MPO) was also decreased in Munc13-4-null mice challenged with CpG (Fig. 1f). Our data indicate that the inflammatory response to in vivo challenge with the eTLR ligand CpG (Fig. 1) but not to the plasma membrane TLR ligand LPS32 is impaired in Munc13-4 deficiency. On the basis of these studies, we next tested the hypothesis that blocking Munc13-4–STX7 binding, therefore, the specific function of Munc13-4 at late endosomes, is a potential approach to counteract endosomal signaling-induced systemic inflammation.

a–d, Inflammatory response to CpG. e,f, Neutrophil number (%) and MPO plasma levels, a marker of neutrophil-mediated systemic inflammation. In a–f, mice were analyzed 6 h after systemic (tail vein) administration of CpG type A (n = 4, WT; n = 7, Jinx) or type B (n = 6, WT; n = 8, Jinx), where n, indicated by each symbol, corresponds to an independent mouse, analyzed in three independent experiments. Data are shown as the mean ± s.e.m. #Outlier (Grubbs test, α = 0.05). NS, not significant. A two-tailed Student’s t-test was used for two-group comparisons.
Source data
High-throughput screen for STX7–Munc13-4 inhibitors
Late endosome-initiated proinflammatory signaling is a central mechanism in systemic inflammation and autoimmunity. We recently showed that this process is directly regulated by the novel interaction between the calcium sensor Munc13-4 and the late endosomal SNARE protein STX7 (ref. 16) (Fig. 2a). The affinity of STX7 for Munc13-4, a calcium sensor, increases in the presence of Ca2+ (ref.16). Expression of a calcium-binding-deficient mutant of Munc13-4 fails to rescue late endosome defects in Munc13-4-deficient cells16. Pharmacological modulators targeting the specific molecular pathways that regulate endosome-initiated inflammation would be beneficial for treating multiple human diseases, including autoimmune and autoinflammatory diseases. To address this clinical need, we developed an assay to screen molecular libraries to identify novel hits that can inhibit the specific binding between Munc13-4 and STX7, two modulators of endolysosomal function necessary for eTLR activation. The assay is based on the principle of FRET from a highly stable fluorescence donor, terbium cryptate (Tb), to EGFP (Fig. 2b and Supplementary Table 1). For the Tb–EGFP pair, the Förster radius is approximately 43 Å (ref. 33). The FRET principle involves energy transfer from a Tb chelate with a long excited-state lifetime to a conventional fluorophore (EGFP) with a short excited-state lifetime. Emission is monitored 100 ms after excitation and acceptor emission is only observed if energy transfer occurs. The time gating eliminates all fluorescence background and less than 1% energy transfer is required for detection, making it a highly sensitive method for monitoring physical interaction34. Notably, in these lanthanide-conjugated antibody-based assays, the conformational flexibility of the antibodies compensates for the more considerable distance between donor and acceptor, providing an abundant opportunity for energy transfer to occur during the extended detection window of the measurement, provided by the prolonged donor-excited-state lifetime33. Our assay uses a Tb-conjugated anti-Flag antibody, which specifically binds to the tag in Flag–Munc13-4 (Fig. 2b). The TR-FRET signal increases following the specific binding of Flag–Munc13-4 to EGFP–STX7 (Fig. 2b). In this assay, the integrity of intracellular organelles is preserved and STX7 localizes at endosomes (Fig. 2b, inset), its natural environment. This ensures that protein presentation recapitulates physiological cellular interactions, increasing the likelihood of finding biologically active compounds. The specificity of the reaction was demonstrated by competitive inhibition using recombinant STX7. Nevertheless, control proteins had no competitive effect (Fig. 2c). This high-throughput screening assay conforms to a ‘mix and measure’ format and is easily automated. The assay is robust, highly reproducible, adaptable to low volumes and liquid-handling devices and has a high signal-to-background ratio (Fig. 2d and Supplementary Fig. 1).

a, Mechanism of STX7 and Munc13-4 regulation of endolysosome signaling, Munc13-4 (orange) binds to STX7 (light green) at the cytosolic side of the late endosome. Munc13-4 is in equilibrium between the free cytosolic and membrane-bound form and specifically binds to membrane-bound STX7 in a calcium-dependent manner. Lysosome fusion to the late endosome forms a degradative compartment where nucleic-acid-sensing TLRs are processed and activated. Specific eTLR ligands lead to the upregulation of proinflammatory cytokines, which are inhibited in Munc13-4 deficiency. b, Schematic representation of the TR-FRET binding assay. Cell lysates expressing Flag–Munc13-4 and GFP–STX7 at late endosomes (LAMP1) (top-left micrograph, representative of three independent experiments) are incubated with Tb-conjugated anti-Flag antibody. The samples are excited at 340 nm. The emission peak of Tb (centered at 490 nm) overlaps with the excitation spectrum of GFP. The FRET signal is measured by detecting GFP emission at 520 nm and results are expressed as the emission ratio of the acceptor (GFP, 520 nm) to donor (Tb, 490 nm, used as internal control). An increased emission ratio is indicative of specific binding. Inset, GFP–STX7 localizes at LAMP1+ endosomes (n = 3). c, Homologous competitive binding assay for STX7 using the TR-FRET assay. Specific binding was measured at a constant [GFP–STX7] (∼50 nM) that was less than half of the IC50 in the presence of various concentrations of recombinant soluble STX7 (aa 1–236). The Kd (∼2.7 µM) was calculated using the homologous competitive binding curve fitted to a built-in equation of one-site competition (n = 3, independent biological samples). Data are shown as the mean ± s.e.m. d, TR-FRET assay. Experiments were carried out using lysates from cells expressing Flag–Munc13-4 and either GFP–STX7 (red) or GFP (black). The calcium-binding-deficient mutant Munc13-4-C2A*B* abolishes signal (blue). Signal in the absence of calcium is shown with green bars. Equimolar expression of GFP and GFP–STX7 was determined by fluorometry (excitation, 488 nm; emission, 520 nm). e, Screenings were performed using the full MBHF4 and MBHF12 libraries (Supplementary Table 1). The library was screened in three independent experiments (representative data are shown). Lysates expressing GFP–STX7 and Flag–Munc13-4 (black) or Flag–Munc13-4-C2 mutant (negative control, red) were used. Compounds (50 nl) were pin-tooled into wells. After the addition of Tb-conjugated anti-Flag antibody, the interactions were started by the addition Ca2+. The TR-FRET signal was read before and 5 min after Ca2+ addition. Compounds found to inhibit Ca2+-induced signal exceeding the 3σ statistical limit (dotted line) are considered initial hits. f, Top hit compounds identified in parent TR-FRET used in follow-up assay and the corresponding percentage inhibition in the TR-FRET assay.
Source data
For the identification of inhibitors of the interaction between Munc13-4 and STX7, we screened the full Maybridge HitFinder 4 (MBHF4) and MBHF12 libraries; the latter has ∼50% compound diversity compared to the MBHF4 (∼15,000 total compounds each). Importantly, the Z′ factor for the full MBHF screen was 0.76 ± 0.08, indicating high sensitivity, reproducibility, robustness and low batch-to-batch and plate-to-plate variability and a hit rate = 0.28% (Fig. 2e and Supplementary Fig. 1). This screening approach led to the identification of ENDOtollins (ENDOs), inhibitors of MUNC13-4–STX7 interaction and eTLR activation. The initial hits were confirmed as inhibitors of Munc13-4–STX7 binding in validation assays and were selected for downstream analysis (Fig. 2f).
ENDO analogs selectively block eTLR endosomal signaling
Next, we studied the effect of the selected small molecules identified in the parent screen on the activation of eTLR9. In this assay, we analyzed the impact of ENDO series on CpG-induced TLR9 activation using cells expressing the human TLR9 gene upstream of an inducible and secretable embryonic alkaline phosphatase (SEAP) reporter system. The assay uses HEK293 cells, which express Munc13-4 and STX7 endogenously, as shown previously by proteomic analysis24 and here by immunoblotting (Supplementary Fig. 2). Stimulating the reporter cells with the TLR9 ligand CpG oligodeoxynucleotide (CpG-ODN) induces the production and secretion of alkaline phosphatase, which is detected in the culture medium by a colorimetric reaction. In Fig. 3a, we show that one of the small molecules that was a positive hit in the parent TR-FRET assay, ENDO3, abolished CpG-mediated activation of TLR9. This compound had minimal effect when tested in HEK293-null cells, which respond to tumor necrosis factor (TNF) using the same SEAP colorimetric reporter system used in HEK-Blue TLR9 cells (Supplementary Fig. 3), thus ruling out nonspecific effects. ENDO7 and, to a lesser extent, ENDO6 also exerted a partial but significant inhibition of TLR9 activation by CpG in this assay (Fig. 3a). On the basis of their performance in TR-FRET assays for the detection of Munc13-4–STX7 binding and their inhibitory activity on TLR9 activation in cell-based assays, we selected ENDO3 and ENDO7 for initial downstream analysis and orthogonal validation.

a, HEK-Blue-hTLR9 reporter cells, expressing the human TLR9 gene and an inducible SEAP reporter gene under the control of the IFNβ minimal promoter fused to five NF-κB-binding and AP1-binding sites, were stimulated with CpG for 24 h (n = 8 independent biological samples representative of three independent experiments). Data are shown as the mean ± s.e.m. A two-way ANOVA with Dunnett’s multiple-comparison test was used to test significance. b, Orthogonal validation of ENDO compounds. Two hits, identified in HTS assays and positive in HEK-Blue cell-based assays (ENDO3 and ENDO7), were further validated in pulldown assays. Both compounds inhibit the STX7–Munc13-4 interaction (10 µM). Data are representative of three independent experiments. c, Cell-based assay for the analysis of TLR9-mediated activation of pDCs. pDCs (cell line CAL1) were incubated with 10 µM ENDO3, ENDO7 or vehicle (DMSO) for 1 h and subsequently stimulated with CpG-ODN or vehicle (n = 3 independent experiments). Data are shown as the mean ± s.e.m. A one-way ANOVA with Tukey’s multiple-comparison test was used to test significance. d, ENDO3 inhibits IRF7 phosphorylation in a time-dependent manner. Inhibition is detected as early as 1 h and maintained through the remaining incubation time. Data are representative of two independent experiments. e, Dose–response study of ENDO3 inhibitor by analysis of CD40 mobilization in Cal1 cells stimulated with CpG. Symbols indicate independent experiments (n = 2). f–l, ENDO3 specifically inhibits activation through CpG but does not inhibit exocytosis through plasma membrane ligands. f, ENDO3 inhibits STX7–MUNC-13-4 binding but is negative in TR-FRET counterscreens using MUNC13-4 and Rab11 or Rab27a (n = 4 biological replicates). Data are shown as the mean ± s.d. A Kruskal–Wallis one-way ANOVA with uncorrected Dunn’s test (two-sided) was used to test significance. g, ENDO3 does not affect the binding of STX7 to the SNARE protein VAMP8 (n = 3 independent biological replicates). Data are shown as the mean ± s.e.m. A two-tailed unpaired Student’s t-test was used to test significance. h, ENDO3 inhibits CpG-induced ERK activation (n = 3 independent experiments). Data are shown as the mean ± s.e.m. A one-way ANOVA with uncorrected Fisher’s least significant difference (LSD) test (two-tailed pairwise comparisons) was used to test significance. i, ENDO3 inhibits CpG-induced CD11b upregulation in neutrophils stimulated with the TLR9 ligand CpG-B (n = 3 for 5 and 20 µM and n = 6 for DMSO and 10 µM, where n indicates the number of independent mice). Data are shown as the mean ± s.e.m. A one-way ANOVA with uncorrected Fisher’s LSD test was used to test significance. MFI, mean fluorescence intensity. j–l, The mobilization of neutrophil secretory organelles in response to fMLF, which activates a plasma membrane receptor, is not affected by ENDO3. j, Mobilization of secretory vesicles (CD11b) (n = 6 independent mice). k, Secondary granules (CD66b) (n = 12 for DMSO and n = 9 for ENDO3, where n indicates the number of independent mice). l, Azurophilic granule exocytosis (MPO) (n = 10 independent mice). CyTD, cytochalasin D; NSt, not stimulated. f, fMLF. In j–l, each symbol represents an independent donor analyzed in three independent experiments. Data are shown as the mean ± s.e.m. A two-tailed unpaired Student’s t-test was used to test significance.
Source data
Munc13-4 interacts with STX7 in a Ca2+-dependent manner16 (Fig. 2). Binding specificity was demonstrated by the inability of Munc13-4 to pull down other STXs or to bind to the fusion regulator Vti1b (ref. 16) according to homologous competitive assays using recombinant STX7 (Fig. 2c) and the use of Munc13-4 inactive mutants (Fig. 2d). Here, the inhibitory effect of ENDO compounds on the binding of Munc13-4 to STX7 was confirmed by an orthogonal validation assay, consisting of the pulldown of Munc13-4 with recombinant STX7. We found that preincubation of lysates expressing Munc13-4 with either the compound ENDO3 or the compound ENDO7 reduced the amount of Munc13-4 detected in the pulldowns compared to controls incubated with vehicle alone (DMSO) (Fig. 3b).
To analyze whether ENDO3 is a specific immunomodulator of TLR9 activation in immune cells, we analyzed the activation of TLR9 in the plasmacytoid dendritic cell (pDC)-like cell line Cal1 (refs. 35,36) stimulated with CpG. As the readout, we studied the upregulation of CD40 at the plasma membrane using high-throughput flow cytometry analysis. Of note, CD40 is a transmembrane glycoprotein surface receptor, a member of the TNF receptor superfamily, and its activation is associated with increased effectiveness of antigen presentation and the upregulation of major histocompatibility complex class II and costimulatory molecules CD80/CD86; thus, the level of CD40 plasma membrane is directly associated with pDC immune responses and autoimmune disease. Here, we show that ENDO3 but not ENDO7 significantly decreases the upregulation of CD40 in response to CpG in pDCs (Fig. 3c), further highlighting the inhibitory activity and biological relevance of this compound against cellular activation by TLR9 ligands. IRF7 is an IFN-inducible transcription factor whose phosphorylation induces homodimerization and activation and whose dysregulation causes autoimmunity. In Fig. 3d, we show that treatment with ENDO3 decreased the detection of phosphorylated IRF7 in Cal1 cells in a time-dependent manner, supporting that the compound inhibits CpG–TLR9 downstream signaling. Lastly, dose–response analysis of the effect of ENDO3 on Cal1 cells indicated an IC50 of 1.17 × 10−7 M (Fig. 3e).
ENDO compounds block TLR9-driven activation but spare exocytosis
In addition to regulating late endosomal maturation through its interaction with STX7, Munc13-4 regulates other critical cellular processes through interactions with the small GTPases Rab27a and Rab11. Thus, Munc13-4 and Rab27a regulate the last steps of lytic and azurophilic granule exocytosis in CTLs and neutrophils, respectively19,25. The Munc13-4–Rab11 interaction regulates the docking of recycling endosomes at the plasma membrane and the production of reactive oxygen species21. To ensure that the compounds identified in our parent and pDC-based assays were specific inhibitors of the interaction between STX7 and Munc13-4 but did not interfere with vesicle docking and exocytosis by inhibiting Rabs–Munc13-4 complexes, ENDO compounds were counterscreened in TR-FRET assays using lysates expressing Flag–Munc13-4 and EGFP–Rab27a or EGFP–Rab11. Similar to the original TR-FRET screen (Munc13-4–STX7), these counterscreens are robust and have excellent signal-to-background ratio21 (Supplementary Fig. 1). In Fig. 3f, we show that ENDO3 specifically inhibited the interaction between Munc13-4 and STX7 but did not inhibit the interactions of Munc13-4 with either Rab27a or Rab11, thus supporting the specificity of ENDO3 toward the endolysosomal function of Munc13-4. We also show that ENDO3 did not interfere with the binding of STX7 to vesicle-associated membrane protein 8 (VAMP8); therefore, it is unlikely to block other STX7-mediated functions (Fig. 3g). These results further validate the approach to target protein–protein interactions rather than specific GTPases or effectors to increase specificity in pharmacological interventions to modulate trafficking pathways.
Our previous studies showed that CpG stimulation induces the rapid activation of neutrophil signal pathways, manifested as increased ERK phosphorylation and marked upregulation of the integrin subunit CD11b at the plasma membrane in an Munc13-4–STX7-dependent manner16. This process is inhibited by chloroquine (CQ) treatment, indicating that CpG signals through the endosomal system16. Contrarily, CD11b upregulation in Munc13-4-knockout (KO) neutrophils was normal in response to the chemotactic peptide fMLF, a physiological stimulus that signals from plasma membrane receptors, further supporting that CD11b plasma membrane upregulation in response to CpG depends on TLR9 activation at endosomes16. We reason that treatment with ENDO3 would recapitulate the Munc13-4-KO phenotype should the inhibitor operate through the inhibition of Munc13-4–STX7 binding. Here, we first demonstrate that, similar to what was shown for Munc13-4-KO neutrophils, treatment with ENDO3 inhibited ERK phosphorylation in wild-type neutrophils stimulated with CpG (Fig. 3h). We further show that, as predicted, treatment with ENDO3 inhibited CpG-dependent CD11b upregulation in neutrophils (Fig. 3i).
The upregulation of neutrophil granule membrane proteins at the plasma membrane depends on the trafficking of intracellular granules, an essential mechanism in the neutrophil innate response. The trafficking and fusion of some of these organelles, including secondary and azurophilic granules, is regulated by Munc13-4 through its interaction with Rab27a (ref. 18) but not by the interaction of Munc13-4 with STX7 (ref. 16). After establishing that ENDO3 does not interfere with Rab27a binding (Fig. 3f), we performed additional cell-based functional assays to rule out a possible off-target effect of ENDO3 in the mobilization of neutrophil granules, a Rab27a-dependent process. To this end, we analyzed the putative impact of ENDO3 on the mobilization of CD11b and CD66b from granules to the plasma membrane and on the secretion of MPO from azurophilic granules using a plasma membrane receptor ligand. Cells were stimulated with the formylated peptide fMLF, which signals through the plasma membrane receptor FRP1 and activates neutrophil secretion independently of endosomal activation or TLR9 function. The assays were performed in the presence of cytochalasin D, which disrupts actin remodeling and facilitates the exocytosis of azurophilic granules. We show that ENDO3 did not inhibit the mobilization of either secretory vesicles (CD11b) (Fig. 3j) or specific granules (CD66b) (Fig. 3k) in response to fMLF stimulation. Furthermore, MPO secretion, a mechanism also regulated by Munc13-4 binding to Rab27a, was not affected by ENDO3 treatment (Fig. 3l) in neutrophils stimulated through plasma membrane receptors, further highlighting the specificity of the inhibitor toward endosomal Munc13-4–STX7 binding and eTLR9 activation by CpG.
ENDO3 blocks endolysosomal flux and in vivo CpG-driven IL-6
Using a quantitative microscopy approach to analyze endolysosomal morphology, we previously showed that Munc13-4 and STX7 regulate endosomal maturation. We also showed that cells lacking Munc13-4 expression have an accumulation of enlarged endosomes. The enlarged LAMP1+ compartment phenotype in Munc13-4-KO was rescued by the expression of Munc13-4 but not by the expression of Munc13-4-C2A*C2B*, a mutant that disrupts its interaction with STX7 (ref. 16). We hypothesize that inhibition of the Munc13-4–STX7 interaction by ENDOs should recapitulate the impaired endosomal maturation phenotype observed in Munc13-4 deficiency, manifested as an enlarged endolysosomal compartment. Here, we show that treatment of cells with ENDO3 for as short as 2 h induced a significant increase in endolysosome size that recapitulated the phenotype observed in Munc13-4-KO cells (Fig. 4a). In addition to enlarged late endosomes, Munc13-4-deficient cells also showed defective late endosomal trafficking37. To directly analyze whether ENDO3 regulates the trafficking of endosomal acidic organelles, we labeled the cells with LysoTracker and quantitatively analyzed organelle dynamics using pseudo-TIRFM (total internal reflection fluorescence microscopy). This technique facilitates the study of organelles that may not necessarily be in areas adjacent to the plasma membrane38. Kinetic analyses showed that acidic organelles in ENDO3-treated cells have impaired trafficking compared to vehicle-treated controls (Fig. 4b and Supplementary Fig. 4), suggesting that, similar to Munc13-4-deficient cells37, ENDO3 decreases endosomal vesicle dynamics. Next, because TLR9 activation requires endosomal maturation and Munc13-4 deficiency was associated with reduced maturation and impaired endolysosomal degradative capacity16, we analyzed the effect of ENDO3 on endolysosomal activity. To this end, we used a cathepsin B substrate derived from cresyl violet (Magic red). This membrane-permeable probe becomes fluorescent upon hydrolysis in endolysosomes, releasing membrane-impermeable fluorescent cresyl violet, which is trapped in endolysosomes. Magic red becomes fluorescent in cathepsin-active endolysosomes but not in cathepsin-inactive terminal lysosomes39. Thus, Magic red fluorescence requires endosomal maturation and the formation of a hybrid endolysosomal compartment. In Fig. 4c, we show that treatment with ENDO3 decreased endolysosomal activity, which recapitulated what was observed in Munc13-4-deficient cells16, further validating that the ENDO compounds inhibit Munc13-4-mediated endosomal maturation. As an additional control, we analyzed a possible negative impact of ENDOs on cell viability. To this end, we studied early apoptosis and cell death using annexin V and propidium iodide, respectively, as described before40. We show that ENDO3 did not cause significant induction of apoptosis or cell death in immune TLR9-expressing human neutrophil-like cells, even at concentrations as high as 40 µM (Fig. 4d). We also show that ENDO3 did not cause cell death in primary neutrophils (Supplementary Fig. 5). Next, to analyze a possible beneficial role for ENDO3 against eTLR-mediated inflammation in vivo, we used a well-established model of CpG-ODN-induced systemic inflammation in mice31. In this model, cytokines are not detected until 4 h of a single intravenous (i.v.) injection of CpG-ODN, a peak of cytokines is detected 6 h after the insult and cytokines levels rapidly decreased to basal levels at 12 h (ref. 31). A mild increase in the number of leukocytes in circulation was detected 6 h after CpG-ODN insult in both vehicle and ENDO3-treated mice (Fig. 4e). Next, we analyzed the effect of ENDO3 on CpG-induced production of IL-6. In Fig. 4f, we show that treatment with ENDO3 significantly prevented the increase in IL-6 plasma levels induced by CpG in mice.

a, ENDO3 inhibits endolysosomal flux. Inhibition of the fusion of late endosomes with lysosomes by ENDO3 results in the enlargement of the LAMP1+ compartment. Left, representative images of 293T cells treated with vehicle or with 10 µM ENDO3 for 2 h. Right, quantification (n = 18 for vehicle, n = 52 for 2 h and n = 100 for 16 h, where n indicates the number of cells). Each symbol represents an individual cell from three independent experiments. Data are shown as the mean ± s.e.m. A one-way ANOVA with Tukey’s multiple-comparison test was used to test significance. b, Analysis of endolysosomal dynamics. Acidic organelles of cells treated with ENDO3 or vehicle were labeled with LysoTracker and analyzed by TIRFM. Vesicle speed from vehicle (DMSO)-treated and ENDO3 (10 µM)-treated cells was segregated in 0.02 µm s−1 increments and plotted as low-speed versus high-speed moving vesicles (b) or in a histogram (Supplementary Fig. 4). The average speed for each speed range is expressed as the mean ± s.e.m. (n = 14 for vehicle and n = 13 for ENDO3, where n indicates the number of cells per condition). A two-tailed unpaired Student’s t-test was used to test significance. c, Representative images (left) and quantification (right) of the analysis of the effect of ENDO3 on functional endolysosomes (cathepsin B activity) using the fluorogenic probe Magic red (n = 21 cells for vehicle and n = 25 cells for ENDO3 from two independent experiments). Data are shown as the mean ± s.e.m. A two-tailed unpaired Student’s t-test was used to test significance. d, Analysis of the effect of ENDO3 on apoptosis (annexin V) and cell death (propidium iodide) in human granulocytes by flow cytometry (n = 6). Data are shown as the mean ± s.e.m. A one-way ANOVA with Tukey’s multiple-comparison test was used to test significance. e,f, ENDO3 decreases IL-6 production in vivo. Mice treated with ENDO3 or vehicle (i.p.) were challenged with a single dose of the TLR9 ligand CpG-B or vehicle and analyzed 6 h after insult. e, Effect of ENDO3 on total white blood cells. f, ENDO3 treatment decreases CpG-mediated inflammation, manifested as significantly decreased IL-6 production (n = 6 mice per group from three independent experiments). Data are shown as the mean ± s.e.m. #Outlier (Grubbs test, α = 0.05). A one-way ANOVA with uncorrected Fisher’s LSD test was used to test significance.
Source data
ENDO12 emerges as a highly potent eTLR inhibitor
On the basis of its performance in the original TR-FRET assay, validation binding assays, orthogonal cell-based assays and counterscreens, we selected ENDO3 for downstream molecular optimization. Using AlphaFold, we identified the most likely protein–protein interaction domains of STX7 and Munc13-4. The interface between Munc13-4 and STX7 comprises the MDH1 domain of Munc13-4 and residues S129–Q148 from the disordered region of STX7 (Fig. 5a). This region is conserved in STX7 through species but it is not present in other STX family members (Supplementary Figs. 6 and 7). Ligand-binding pocket prediction for the AlphaFold v3 complex using Discovery Studio version 21.1.0.20298 identified the pocket at the interface between the Munc13-4 MDH1 domain and the STX7 disordered region as the top-scoring binding site (Supplementary Fig. 6d). This finding was validated by mutagenesis analysis. We show that replacing the R140, N141, L142 and W145 residues with alanine or deleting residues 129–148 of the disordered linker region of STX7 decreased binding to Munc13-4 in TR-FRET assays (Fig. 5b). This modification in the disordered region of STX7 reduced the colocalization of STX7 with Munc13-4 (Fig. 5c), further validating the importance of the STX7 disordered region in a cell-based assay.

a, The Munc13-4 (light blue)–STX7 (yellow) complex according to AlphaFold v3 (10.24.23) identifies a molecular interface formed by the disordered region of STX7 and the MDH1 domain of Munc13-4 (right, magnified view). Inset, ChimeraX analysis of the protein–protein interaction interface of Munc13-4 (light blue) and STX7 (yellow) identifies residues R140, N141, L142 and W145 in STX7 as the amino acids with the highest likelihood of interaction (dark blue solid lines) with MUNC13-4. The corresponding PAE plot is presented in Supplementary Fig. 6. b, TR-FRET analysis of the binding of Munc13-4 to STX7, its quadruple mutant R140A;N141A;L142A;W145A (RNLW-A) or the deletion mutant Δ129–148 (n = 5 independent biological replicates). Data are shown as the mean ± s.e.m. A one-way ANOVA with uncorrected Fisher’s LSD test was used to test significance. c, Expression of STX7 lacking the molecular interface (Δ129–148) decreases its association with Munc13-4 in living cells (n = 4 independent experiments). Data are shown as the mean ± s.e.m. A two-tailed paired Student’s t-test was used to test significance. d, ENDO3 analogs tested in downstream assays. The predicted affinity and effective inhibitory activity of all ENDO analogs are presented in the associated Supplementary Table 2. e, Analysis of SAR-derivative compounds in cell-based TLR9 activation assays. Compounds were tested using the HEK-Blue reporter cell-based assay. In this analysis, cells were treated with the indicated compound or vehicle for 1 h before the addition of the TLR9 ligand CpG (n = 8 independent biological replicates). ENDO3 and ENDO12 are denoted by the red bars. Data are shown as the mean ± s.e.m. A one-way ANOVA with Dunnett’s multiple-comparison test was used to test significance. f, Analysis of the effect of the ENDO series on IRF signaling. IRF reporter cells were treated with the indicated compound or vehicle (DMSO) for 1 h and subsequently stimulated with the TLR3 agonist poly:IC (n = 3 independent biological replicates). Data are shown as the mean ± s.e.m. A one-way ANOVA followed by Dunnett’s multiple-comparison test was used to test significance. g, Model representing the interaction of ENDO3 and ENDO12 in the pocket identified in a. Several residues of STX7 and Munc13-4 within 3 Å of the compounds are highlighted. This includes R140 in the interface shown in a. The predicted hydrogen bond formed by ENDO12 (but not ENDO3) with S137 in the disorder region of STX7 is indicated with small pink dots. h, SPR analysis of ENDO12 binding to recombinant STX7. Top, representative binding curves from three independent experiments. Bottom, individual and average KD values for ENDO3 and ENDO12 binding assays (n = 3). i, Dose–response analysis of the effect of the indicated compounds on TLR9 activity (n = 3 independent biological samples). Data are shown as the mean ± s.e.m. j–l, Immunofluorescence analysis of endogenous proteins in RAW 264.7 murine macrophages. Where indicated, the cells were treated with ENDO3, ENDO12 (10 µM) or vehicle (DMSO, NSt) for 1 h before analysis. j, ENDO12 significantly decreases the colocalization of STX7 with Munc13-4. k,l, ENDO12 decreases the colocalization of TLR7 (k) and TLR9 (l) at LAMP1+ organelles (n = 3 (j,l) or n = 4 (k), where n indicates the number of independent experiments). Data are shown as the mean ± s.e.m. Statistical analysis of Superplots was calculated on the basis of the average values of each experiment (large symbols). A repeated-measures one-way ANOVA with uncorrected Fisher’s LSD test was used to test significance.
Source data
Next, to generate ENDO3 analogs, we used several independent SAR strategies. We replaced the ENDO3 pyridine ring, azo moiety, bromine and the hydroxyl group with alternative fragments (Maybridge) (Fig. 5d). Molecular docking studies indicated that many of these analogs maintain high-affinity constants and engage the binding pocket in the Munc13-4 MDH1–STX7 disordered region interface (Supplementary Table 2). Next, SAR-derivative compounds were synthesized and vetted in functional assays consisting of the analysis of TLR9 activation using the HEK-Blue reporter cell line described above in Fig. 3. In Fig. 5e, we show that ENDO3 and ENDO12 (red bars) but not other derivatives exert a potent inhibitory activity on TLR9 activation. In an additional cell-based assay for the analysis of eTLR activation, we analyzed the ability of ENDO analogs to inhibit IRF signaling stimulated with the TLR3 agonist poly:IC in Jurkat cells (Fig. 5f). ENDO3 and ENDO12 were the most potent inhibitors in this assay, further supporting their roles as specific inhibitors of eTLR activation (Fig. 5f). ENDO4, ENDO5, ENDO11 and ENDO13 also showed inhibitory activity in this assay. According to the SAR study, replacing ENDO3 pyridine ring with a heterocyclic scaffold, 1H-imidazo[4,5-b] pyrazine (ENDO14) or rearrangement of the bromine and methyl substituents on the pyridine ring (ENDO17, ENDO18 and ENDO19) did not improve activity. Further replacement of the azo group with ethylene (ENDO16, ENDO17 and ENDO19), vinylene (ENDO14 and ENDO18) or pyrazine (ENDO15) and or of the C11 hydroxyl group with methoxy (ENDO18) did not result in improved activity. Replacing the bromine with a hydroxyl group (ENDO13) and introducing a phenylacetamide (ENDO11) at the diethylamino end of ENDO3 did not result in activity improvement (Fig. 5e,f). The two most active ENDOs, ENDO3 and ENDO12, are molecularly and structurally similar (Fig. 5d). However, in ENDO12, a fluorine group, generally considered more amenable for downstream medicinal chemistry applications than bromine41, replaces the bromine group present in ENDO3. Both ENDO3 and ENDO12 are predicted to fit in the pocket formed by the STX7–Munc13-4 interface (Fig. 5g) and are predicted to bind R140; however, only ENDO12 forms a hydrogen bond with S137 in the disordered region of STX7. Surface plasmon resonance (SPR) analysis showed that both ENDO3 and ENDO12 bind to STX7, although ENDO12 binds with higher affinity (ENDO3: 3.1 ± 0.7 µM and ENDO12: 2.7 ± 0.7 µM, n = 3; Fig. 5h). In agreement with this, ENDO12 showed increased inhibitory activity in cell-based assays compared to ENDO3 (Fig. 5i). Thus, dose–response analyses confirmed that ENDO12 was the most active compound with an IC50 of 1 × 10−7 M (Fig. 5i). Despite this difference, both ENDO3 and ENDO12 decreased the colocalization of STX7 with Munc13-4 in living cells, validating their mode of action in intact cells (Fig. 5j). Both compounds also decreased the colocalization of TLR9 with LAMP1 (Fig. 5k), supporting that the interaction of Munc13-4 with STX7 is necessary for the maturation of the TLR9 compartment. Lastly, ENDO12 significantly decreased the colocalization of TLR7 with LAMP1 (Fig. 5l) but neither compound significantly altered the colocalization of TLR7 or TLR9 with Rab5+ early endosomes or Rab7+ late endosomes (Supplementary Fig. 8).
ENDO12 reduces inflammation but spares antiviral response
CpG-ODN-initiated TLR9 signaling mediates the release of IL-6 and type I IFN-mediated response, which, if exacerbated, leads to systemic inflammation and contributes to autoimmunity but does not recapitulate a fully antiviral response, which involves both eTLR-dependent and independent mechanisms. Here, to understand the ability of ENDOs to inhibit eTLR activation, we first isolated mouse primary splenic CD11c+ DCs and analyzed their response to TLR3, TLR7 and TLR9 ligands ex vivo. We found that the activation of primary DCs by CpG, denoted by the surface expression of CD40, was inhibited by ENDO12 and, to a lesser extent, by ENDO3 (Fig. 6a). The plasma membrane mobilization of CD40 in DCs stimulated with the TLR7 ligand CL097 was significantly inhibited by ENDO12 but not by ENDO3 (Fig. 6a). Poly:IC (TLR3) did not significantly stimulate the mobilization of CD40. In an orthogonal assay, we investigated the effect of these eTLR agonists on the production of IL-6 and IFNα by splenic CD11c+ DCs. Of note, CpG and CL097 but not poly:IC stimulated IL-6 secretion in this assay, while poly:IC was the most efficient stimulus in inducing IFNα secretion by CD11c+ DCs (Fig. 6b,c). We found that ENDO12 significantly inhibited IL-6 production following CpG or CL097 stimulation (Fig. 6b). IFNα production was also inhibited by ENDO12 when stimulated with eTLR ligands (Fig. 6c).

a–c, Spleen-isolated primary DCs were treated with ENDO12 or ENDO3 followed by stimulation with the eTLR ligands CpG (TLR9), CL097 (TLR7) and poly:IC (TLR3) for 48 h. a, CD40 surface expression was analyzed by flow cytometry (n = 3 mice per condition). Data are shown as the mean ± s.e.m. A repeated-measures two-way ANOVA with uncorrected Fisher’s LSD test was used to test significance. b,c, IL-6 (b) and IFNα (c) secretion by CD11c+ DCs (n = 5 mice per condition analyzed in two independent experiments). Data are shown as the mean ± s.e.m. A repeated-measures one-way ANOVA with uncorrected Fisher’s LSD test was used to test significance. d–f, Effect of ENDO12 in systemic inflammation. Mice treated with ENDO12 or vehicle (i.p.) were challenged with a single dose of the TLR9 ligand CpG-A-ODN or vehicle and IL-6 (d), IFNγ (e) or MPO (f) were analyzed 6 h after insult. In d–f, each symbol represents an independent mouse (n = 7 (ENDO12) and n = 8 (vehicle) mice per condition). Data are shown as the mean ± s.e.m. A one-way ANOVA with uncorrected Fisher’s LSD test was used to test significance. g–l, ENDO12 does not inhibit the in vivo response to LCMV infection. g–k, Cytokine plasma levels of mice treated with ENDO12 (i.p. 15 mg kg−1) or vehicle (5% DMSO in PBS) and infected with LCMV or treated with PBS as a control were analyzed by multiplex technology. MPO was analyzed by ELISA (l). In g–l, Each symbol corresponds to an independent mouse (n = 6 mice per condition). Data are shown as the mean ± s.e.m. A one-way ANOVA with uncorrected Fisher’s LSD test was used.
Source data
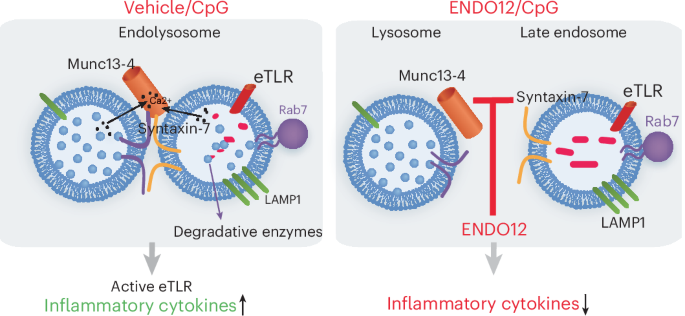
Next, we analyzed the effect of ENDO12 on eTLR-mediated inflammation in vivo, using a mouse model of CpG-ODN-induced systemic inflammation. Here, we show that in vivo secretion of the proinflammatory mediators IL-6 and IFNγ in response to CpG was significantly decreased in the plasma of CpG-challenged mice after treatment with ENDO12 (Fig. 6d,e). We also found that treatment with ENDO12 significantly decreased neutrophil secretion of azurophilic granule cargoes (MPO) in response to CpG (Fig. 6f), an effect that, as shown in Fig. 3 for the compound ENDO3, is independent of the Rab27a-mediated Munc13-4 function in secretion but is instead mediated by the impact of ENDOs on the STX7–Munc13-4 interaction regulating CpG-mediated endosomal activation. These studies highlight ENDOs, particularly ENDO12, as new inhibitors of the endosomal function of Munc13-4, demonstrating pharmacological complementation of the genetic phenotype, and support the notion that ENDO12 is a potent inhibitor of eTLR both ex vivo and in vivo.
Lastly, we investigated the potential impact of ENDO12 on the host response to viral infection. In these assays, we used the lymphocytic choriomeningitis virus (LCMV) model of infection, a virus that activates both eTLR-dependent and independent mechanisms42. We show that the production of cytokines with antiviral activity, including IL-6, IFNα and IFNγ, was not affected by ENDO12 (Fig. 6g–i). ENDO12 only mildly decreased the production of the macrophage inflammatory proteins MIP1β and MIP2 (Fig. 6j,k) in this viral infection model. All other cytokines and chemokines studied were not affected by ENDO12 treatment (Supplementary Fig. 9). LCMV infection triggered a marked systemic neutrophil response, characterized by increased plasma levels of azurophilic granule cargoes. This neutrophil response was also not affected by ENDO12 (Fig. 6l). Altogether, our data support that ENDO12 is a significant inhibitor of eTLR activation and mediates anti-inflammatory activity in vivo in the setting of eTLR-mediated inflammation. However, ENDO12 does not interfere with the host’s response to viral infection.