
Mice and cell line
Rnf213 knockout (Rnf213–/–, KO) mice on a C57BL/6 background were generously provided by Xiaopeng Qi. Spib knockout (Spib–/–) mice on the Rnf213–/– background were generated by Yinming Liang. Briefly, a sgRNA was designed to result in the complete deletion of exon 2 (approximately 144 bp) in the Spib gene upon CRISPR/Cas9-mediated cleavage. Rnf213–/–Spib+/– mice were then crossed to obtain Rnf213–/–Spib–/– (DKO) mice. C57BL/6 (CD45.1+) mice were obtained from Charles River (Beijing, China). All the mice used in the experiments were aged 6 to 8 weeks and housed under specific pathogen-free (SPF) conditions, with a 12 h light/dark cycle and ad libitum access to food and water. The study was conducted in accordance with the national guidelines for animal research and was approved by the Animal Experiment Ethics Committee of Tongji Medical College (Protocol IACUC-4839). All methods were carried out in compliance with relevant regulations and ethical standards. HEK293T cells were generously provided by Hongmei Yang.
Cell isolation and purification
The cells were extracted separately and resuspended in red blood cell lysis buffer (Beyotime, C3702). The single-cell suspensions were subsequently filtered for flow cytometry. Splenocytes were isolated via Ficoll-Paque PLUS (Cytiva, 17144003) density gradient centrifugation. Afterward, the T cells were depleted by incubation with Guinea Pig Complement (Rockland Immunochemicals, C300-0500) and anti-Thy-1.2 (BioLegend, 105301) for 30 min at 37 °C. Subsequently, purified splenic B cells were obtained by culturing the cells in a surface-treated T75 flask and maintaining them in an incubator (37 °C, 5% CO2) for 1 h, during which the monocyte cells were removed.
Flow cytometry and cell sorting
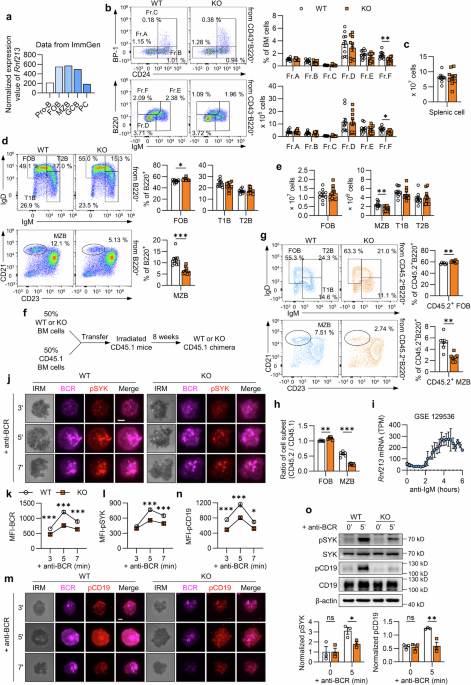
To analyze the various B-cell subsets in mice, the cells were stained with distinct surface and intracellular antibodies. Splenocytes were stained with BV510-anti-B220 (BioLegend, 103248), FITC-anti-CD19 (BioLegend, 115506), PE-anti-CD23 (BioLegend, 101608), APC-anti-CD21 (BioLegend, 123412), PE/Cy7-anti-CD93 (BioLegend, 136506), APC/Cy7-anti-IgD (BioLegend, 405716), BV421-anti-IgM (BioLegend, 406518), PE-anti-BAFFR (BD Biosciences, BD, 565783), FITC-anti-CD95 (BD, 554257), AF647-anti-GL7 (BioLegend, 144606), PE-anti-CXCR4 (BioLegend, 146506), PB-anti-CD86 (BioLegend, 105022), APC-anti-CD45.1 (BD, 558701), BV510-anti-CD45.2 (BD, 740131), and PE-anti-NP (Biosearch, N-5070) antibodies. To classify B cells in the BM, the cells were labeled with BV510-anti-B220, APC-anti-CD43 (BioLegend, 143208), PE-anti-BP-1 (BioLegend, 108307), PE/Cy7-anti-CD24 (BioLegend, 101822), and BV421-anti-IgM antibodies. Peritoneal cells were stained with BV510-anti-B220, FITC-anti-CD19, APC-anti-CD43 and RB780-anti-CD5 (BD, 755635) antibodies. Additionally, BV605-anti-Ki-67 (BioLegend, 652413) or PE/Cy7-anti-Ki-67 (BioLegend, 151218) was used to label proliferating cells; BV605-Annexin Ⅴ (BD, 563974) or PE-Annexin Ⅴ (BioLegend, 640908) was used to detect apoptotic cells; and 7-AAD viability staining solution (BioLegend, 420404) was used to distinguish live and dead cells.
To minimize nonspecific binding, the cells were blocked with anti-CD16/CD32 (BioLegend, 101319) antibodies on ice for 30 min prior to antibody staining. For surface staining, the cells (5 × 105) were stained with surface antibodies diluted in PBS containing 2% fetal bovine serum (FBS) (Vazyme, F101-01) for 30 min on ice. Intracellular staining (5 × 105) was carried out after fixation and permeabilization by the Foxp3 Fixation/Permeabilization Concentrate and Diluent (Invitrogen, 00-5521-00). The cells were then incubated with intracellular antibodies for 30 min on ice. Sample data were measured via an Attune™ NxT flow cytometer (Thermo Fisher), and the data were processed with FlowJo software (Tree Star).
For splenic B-cell sorting, splenocytes were labeled with FITC-anti-B220 (BioLegend, 103206) and then sorted on a FACSAria™ III sorting flow cytometer (BD).
Western blot
Purified splenic B cells (3 × 106) were incubated with 5 μg/mL biotin-F(ab’)2-anti-Ig (M + G) (anti-BCR, Jackson ImmunoResearch, 115-066-068) on ice, followed by incubation with 20 μg/mL streptavidin (Jackson ImmunoResearch, 016-000-114) on ice. The cells were incubated at 37 °C for 5 min, and the reaction was terminated with precooled PBS. For anti-CD40 or IL-4 activation, purified B cells (3 × 106) were activated with 15 μg/mL anti-CD40 (SelleckChem, A2136) or 20 ng/mL IL-4 (GenScript, Z02996) at 37 °C for 15 min. Subsequently, cell lysis was carried out with RIPA lysis buffer (Beyotime, P0013B). The cell lysates were examined via SDS-PAGE, and immunoblotting was performed with the following antibodies: anti-pSYK (Cell Signaling Technology, CST, 2710S), anti-SYK (CST, 13198S), anti-pCD19 (CST, 3571S), anti-CD19 (CST, 90176S), anti-β-actin (Proteintech, 66009-1-Ig), anti-pNFκB pP65 (CST, 3033S), anti-NFκB P65 (CST, 4764S), anti-pSTAT1 (CST, 9167S), anti-STAT1 (CST, 14994S), anti-pSTAT3 (CST, 9145S), anti-STAT3 (CST, 9139S), anti-pSTAT5 (CST, 4322S), anti-STAT5 (CST, 94205S), anti-pPI3K pP85/pP55 (CST, 4228 L), anti-PI3K P85 (CST, 4292S), anti-pAKT (CST, 4060 L), anti-AKT (CST, 9272S), anti-pmTOR (CST, 5536S), anti-mTOR (CST, 2983S), anti-pS6 (CST, 4858 L), anti-S6 (CST, 2217S), anti-pFOXO1 (CST, 9461S), anti-FOXO1 (CST, 2880S), anti-CTCF (ABclonal, A19588), anti-SPIB (CST, 14337S), anti-ETS1 (ZenBio, R382074), anti-GABPA (HuaBio, HA500187), anti-FLI1 (HuaBio, HA500149), anti-PU.1 (ABclonal, A20461), anti-GAPDH (Proteintech, 60004-1-Ig), anti-PIK3C3 (ABclonal, A12295), anti-PTEN (CST, 9188S), anti-pSHIP-1 (CST, 3941S), SHIP-1 (ABclonal, A3571), anti-NOTCH2 (ABclonal, A24877), anti-pHSL (CST, 4126S), anti-HSL (CST, 4107S), anti-ATGL (CST, 2138S), anti-LC3 Ⅰ/Ⅱ (ABclonal, A5618), IκBα (ABclonal, A19714), pIκBα (SelleckChem, F2237), pIKKα/β (CST, 2697S), pP38 (SelleckChem, F0159), HSP90 (ABclonal, A5027), pJNK1/2 (ABclonal, AP0473), pERK1/2 (ABclonal, AP0472).
Bone marrow chimeras
Chimeric mice were established with intravenous injection of a 1:1 mixture of bone marrow cells derived from C57BL/6 (CD45.1+) donors and from either WT or KO (CD45.2+) donors into irradiated (7 Gy) CD45.1+ recipients. After 8 weeks, splenic cells from the recipient mice were extracted and analyzed by flow cytometry.
Measurement of ROS, the mitochondrial membrane potential and BODIPY
For ROS and mitochondrial membrane potential measurements, purified splenic B cells were stimulated with 5 μg/mL anti-BCR for 3 h at 37 °C and 5% CO2. For BODIPY measurement, purified splenic B cells were stimulated with LPS (Sigma, L2880), anti-CD40, CpG (InvivoGen, tlrl-1826-1) or anti-BCR for 24 h and 48 h. The cells were subsequently incubated at 37 °C for 15 min with a mixture of BV510-anti-B220 and Fixable Viability Stain 700 (BD, 564997), which contained either CellROX Green Reagent (Invitrogen, C10444), PK Mito Red (Genvivo, PKMR-1/2), or BODIPY (Invitrogen, D3922). Flow cytometry was performed to evaluate the cellular ROS levels, the mitochondrial membrane potential and the BODIPY fluorescence intensity.
Immunofluorescence
Confocal microscopy was used to examine the expression patterns of PI3P, PIP3, PTEN, and EEA1. Purified splenic B cells (2 × 105 per well) were stimulated with Alexa Fluor 594-F(ab’)2-anti-IgM+IgG (H + L) (Jackson ImmunoResearch, 115-586-068) for 0 and 5 min. The cells were then fixed with 4% paraformaldehyde and permeabilized with 0.05% saponin buffer (Sigma, S4521-10G). The cells were subsequently stained with primary antibodies followed by the corresponding secondary antibodies and DAPI (Beyotime, C1005). The following antibodies were used: anti-EEA1 (HuaBio, HA722147), anti-PTEN (HuaBio, RT1519), anti-PI3P (Echelon Biosciences, Z-P003), anti-PIP3 (Echelon Biosciences, Z-P345B), Alexa Fluor 647 goat anti-rabbit IgG (Thermo, A-21245), and Alexa Fluor 488 goat anti-mouse IgG (Thermo, A-11001). All procedures were performed under protection from light. A Nikon confocal microscope was used to capture the images. Data processing was performed by NIS-elements AR 5.01 software. Each dataset was composed of analyses from over 40 randomly selected individual cells.
Total internal reflection fluorescence microscopy (TIRFm) was employed to visualize the activation of pCD19 and pSYK in response to membrane-tethered antigen stimulation, as described previously.68 Briefly, purified splenic B cells (6 × 105 per well) were incubated with Alexa Fluor 647-Fab-anti-IgM (Jackson ImmunoResearch, 115-607-020) and then activated with an anti-BCR-tethered lipid bilayer at 37 °C for 3, 5, or 7 min in coverslip chambers (Nalge Nunc International). The cells were then fixed with 4% paraformaldehyde and permeabilized with 0.05% saponin buffer. The cells were subsequently stained with primary antibodies, followed by incubation with the appropriate secondary antibodies. The following antibodies were used: anti-pSYK, anti-pCD19, and Alexa Fluor 546 goat anti-rabbit IgG (Thermo, A-11010). All the above procedures were performed in the dark. Images were taken on a Nikon TIRFm. Interference reflection microscopy (IRM) was utilized to determine the B-cell contact area. The MFI in the contact zone was quantified via NIS-Elements AR 5.01 software. Each dataset was composed of analyses from over 30 randomly selected individual cells.
Calcium assay following BCR stimulation
Purified splenic B cells (2 × 106 per well) were washed twice with calcium-free HBSS (Servicebio, G4203) to remove FBS and resuspended in calcium-free HBSS containing 0.5 μM Fluo-4 AM (Beyotime, S1060) and APC-anti-B220 (BD, 553092). Subsequently, the cells were seeded into the polylysine-coated chamber for incubation at 37 °C for 30 min. Following the removal of excess dye, prewarmed RPMI-1640 medium (Servicebio, G4535) was added to the chamber. Following 30 s of baseline fluorescence recording, the cells were stimulated with prewarmed anti-BCR (5 μg/mL final concentration). The fluorescence intensity was continuously monitored by a Nikon confocal microscope and analyzed by NIS-elements AR 5.01 software.
Seahorse metabolic assay
Purified splenic B cells were cultured with 5 μg/mL anti-BCR for 8 h at 37 °C with 5% CO2. After being washed, the cells were resuspended in Seahorse XF Base Medium (Agilent, 102353-100) and seeded into XFe24 plates (Agilent, 102342-100) at 2 × 106 per well. The following reagents were applied to the probe plates for the oxygen consumption rate (OCR) measurement: 1.5 μM oligomycin (Absin, abs42024304), 1 mM FCCP (Sigma, C2920), and 500 nM rotenone (Sigma, R8875) plus 1 μM antimycin A (Sigma, A8674). The extracellular acidification rate (ECAR) was measured by sequentially adding 10 mM glucose (Sigma, G8769), 2 μM oligomycin, and 5 mM 2-deoxy-D-glucose (Sigma, D8375). Finally, the cells were analyzed via a Seahorse XFe24 Cell Metabolism Analyzer (Agilent), and the values were normalized to the absolute cell number in each well.
RNA-seq
Two hundred nanograms of total RNA was submitted to BGI Genomics for library preparation and sequencing. Briefly, total RNA was subjected to mRNA enrichment, fragmentation, and cDNA library preparation. The libraries were adapter-ligated, amplified, and sequenced on a paired-end platform. The raw reads were processed by the proprietary mRNA-seq analysis pipeline of BGI Genomics. The DESeq2 (V1.36.0) package69 was used to identify differentially expressed genes (DEGs). The resulting DEGs were visualized via unsupervised clustering and heatmaps with the pheatmap package. Gene set enrichment analysis (GSEA) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were carried out with the clusterProfiler package (V4.7.1.003).70 The gene expression profile was first screened with an average FPKM > 0.1 to rule out genes with low expression, and significantly altered genes were then defined as those with a |log₂ fold change | > 0.5 and P < 0.05.
ATAC-seq
ATAC-seq was performed as previously described.71 Briefly, a total of 5 × 104 sorted splenic B cells were lysed, and the nuclei were tagmented using the TruePrep DNA Library Prep Kit V2 (Vazyme, TD501). Libraries were subsequently subjected to paired-end sequencing at Annoroad Gene Technology. The analysis pipeline was adapted from an established method.72 Specifically, adapter-trimmed reads were aligned to the mm10 genome via Bowtie2 (V2.4.0).73 Broad chromatin peaks were identified with Macs2 (V2.2.7),74 and differentially accessible peaks were called by DESeq2 (V1.36.0)69 with a threshold of |log₂ fold change | > 0.5 and P < 0.05. The broad peak profile was first filtered to exclude regions with average reads ≤ 1. Motif enrichment and peak annotation were performed with HOMER (V4.11).75 The reads were normalized to the library size, and browser tracks were visualized with the IGV browser (V2.8.2).76 To integrate the chromatin accessibility data with histone modification landscapes (H3K4Me1, H3K4Me3, and H3K27Ac, GSE150495), a metagene analysis was performed by Deeptools2 (V3.3.2).77 For footprint analysis, SPIB footprints were scanned and called by HINT-ATAC.78 Quality control of ATAC-Seq data was performed with Ataqv (V1.1.1).79
CUT&Tag and analysis
CUT&Tag was performed as previously described.71 Briefly, sorted splenic B cells (1 × 105) were processed with a Vazyme kit (TD904) according to the manufacturer’s instructions. The cells were first washed and bound to activated ConA beads. Then, the cell-bead complexes were incubated with anti-SPIB at 4 °C overnight, followed by incubation with secondary antibody at room temperature for 1 h. After washing, the samples were incubated with pA/G-Tnp Pro under identical conditions. For DNA fragmentation, the supernatant was discarded, replaced with the indicated buffer, and the mixture was incubated at 37 °C for 1 h. The reaction was stopped by adding 10% SDS and spike-in DNA (1 pg for 105 cells), followed by incubation at 55 °C for 10 min. After this, the DNA was extracted from the supernatant for PCR amplification. Finally, the purified PCR products were sequenced by Annoroad Gene Technology.
For CUT&Tag analysis, adapter-trimmed reads were aligned to the mm10 genome. Subsequent processing followed an established CUT&Tag workflow (Ye Zheng, https://yezhengstat.github.io/CUTTag_tutorial/) to generate a peak matrix. Peak calling was performed using a method analogous to ATAC-seq analysis. The peak matrix and BigWig files for IGV browser visualization were all normalized to the spike-in DNA.
Plasmid construction and Co-IP
The coding sequence of mouse SPIB was derived from GenBank (NCBI reference sequence: NM_019866.1, CCDS21209.1). The Flag-tagged SPIB was amplified by the following primers: 5’-CCCAAGCTTATGGACTACAAGGACGACGATGACAAGATGCTTGCTCTGGAGGCTGCAC-3’ (forward) and 5’-GCTCTAGATCAGACATGCCGGGAG-3’ (reverse). The amplified DNA fragments were subsequently cloned and inserted into the pcDNA3.1 vector by HindIII (NEB, R3104S) and XbaI (NEB, R0145S) dual digestion, followed by T4 ligation (Takara, 2011B). The plasmids expressing V5-tagged mouse RNF213 or RNF213 W3974R were kindly provided by Xiaopeng Qi. All the plasmids generated in this study were verified via DNA sequencing. Lipomaster 3000 Transfection Reagent (Vazyme, TL301-01) was used for transient transfection into HEK293T cells according to the manufacturer’s instructions.
For immunoprecipitation (IP), HEK293T cells were collected 40 h post transfection and lysed with IP buffer containing 2 mM EDTA, 50 mM Tris-HCl [pH 7.4], 150 mM NaCl, and 1% NP-40. The supernatant was collected and incubated with 3 μg of anti-V5-Tag (ABclonal, AE017) and BSA-blocked protein A/G PLUS-agarose (Santa Cruz, sc-2003) for 12 h. Subsequently, the samples were washed with IP buffer and boiled in SDS loading buffer. Both IP samples and input lysates were examined by SDS-PAGE, and immunoblotting was performed with anti-V5-Tag and anti-SPIB antibodies.
Protein stability measurement
For protein stability measurement, pCDNA3.1-Flag-SPIB was co-transfected into HEK293T cells together with pCDNA3.1-V5-RNF213 or empty pCDNA3.1 as a control. The cells were treated with 20 μg/mL cycloheximide (MCE, HY-12320) for 2, 4 or 8 h in the presence or absence of 20 μM MG132 (MCE, HY-13259) or 100 nM bafilomycin A1 (TargetMol, T6740) at each time point and harvested at predetermined time intervals. The cells were subsequently lysed with RIPA lysis buffer, and the lysates were analyzed via Western blotting.
Ubiquitylation assay
Ubiquitin-expressing plasmids were kindly provided by Xiaopeng Qi. For exogenous ubiquitylation detection, plasmids expressing Flag-SPIB, V5-RNF213 or V5-RNF213 W3974R, HA-ubiquitin (WT) or HA-ubiquitin (K6, K11, K27, K29, K33, K48, K63 or K11R ubiquitin mutant) were transfected into HEK293T cells with 20 μM MG132. For endogenous analysis, purified splenic B cells were incubated with 5 μg/mL anti-BCR, followed by incubation with streptavidin on ice. B cells were thereafter stimulated at 37 °C for 30 min. Transfected HEK293T cells and treated splenic B cells were lysed in lysis buffer containing 1.5% SDS and 50 mM Tris-HCl [pH 6.8] and subsequently boiled. Finally, the IP samples and input lysates were subjected to immunoprecipitation with anti-V5-Tag, anti-SPIB, or anti-DDDDK-Tag (ABclonal, AE092), anti-Ubiquitin (PTM BIO, PTM-1107), or anti-HA-Tag (ABclonal, AE008) antibodies.
PI3P and PIP3 measurements
Splenic B cells were labeled with FITC-conjugated anti-B220 and resuspended in PBS containing 5% FBS plus either 1 μM SAR405 (MCE, HY-12481) or DMSO (MP Biomedicals, 196055). After a resting period at 37 °C for 15 min, pre-warmed 5 μg/mL anti-BCR, also containing DMSO or SAR405, was added to the cells. Stimulation was carried out for 0, 5, 10, or 30 min and terminated with 4% paraformaldehyde. Subsequently, cell fixation and permeabilization were performed with the Foxp3 Fixation/Permeabilization Concentrate and Diluent. Intracellular staining was performed with anti-PIP3 or anti-PI3P antibodies, followed by incubation with Alexa Fluor 405 goat anti-mouse IgG (Thermo, A-31553).
Immunizations and ELISA
For T-dependent immunization, 8-week-old WT and KO mice were intraperitoneally injected with 40 μg of NP-KLH (Biosearch, N-5060-25) emulsified in an Alhydrogel adjuvant (InvivoGen, vac-alu-50). Four weeks after the primary immunization, the serum samples were harvested, and the mice received a booster injection of the same formulation. Five days later, splenocytes were isolated for flow cytometry, and serum samples were subjected to ELISA. In addition, 40 μg of NP-Ficoll (Biosearch, F-1420-100) emulsified with Aalhydrogel adjuvant was used for T-independent immunization. One week after immunization, splenic lymphocytes and sera were collected for subsequent flow cytometry or ELISA.
To assess the NP-specific IgM, IgG, and IgG1 levels in immunized mice, 96-well plates were coated with 2 μg/mL NP2-BSA (Biosearch, N-5050XL) or 2 μg/mL NP29-BSA (Biosearch, N-5050H) and blocked with BSA. Subsequently, the diluted serum samples were seeded and incubated at 37 °C for 1 h. NP-specific IgM, IgG, and IgG1 were detected via HRP-goat anti-mouse IgM (Bethyl Laboratories, A90-101P), HRP-goat anti-mouse IgG (Absin, abs20001SS), and HRP-goat anti-mouse IgG1 (Bethyl Laboratories, A90-105P), respectively. After washing, TMB substrate reagent (BD, 555214) was added, and the reaction was terminated with 10% sulfuric acid. The absorbance at 450 nm was measured by a BioTek Synergy H1 Multimode Reader.
PIK3C3 inhibition in vitro and in vivo
For in vitro inhibition, cells were pretreated with 1 μM SAR405 at 37 °C for 1 h, with DMSO used as the control.
For in vivo treatment, 8-week-old WT and KO mice were intraperitoneally injected daily with 5 mg/kg SAR405 dissolved in DMSO for 30 days, while control mice received DMSO.
Statistical analysis
Statistical significance was evaluated with GraphPad Prism (v10.1.2). Student’s t-tests were employed for comparisons between two independent groups. One-way ANOVA or Two-way ANOVA followed by Tukey’s multiple comparisons tests were used for comparisons involving more than two datasets. The following symbols indicate statistical significance: *P < 0.05, **P < 0.01, ***P < 0.001; ns, not significant. Details regarding the statistical tests used for comparative analyses are provided in the legends for the relevant figures.