
Fish husbandry and samples collection
All the killifish used in this study are from the GRZ strain. We housed fish in plastic tanks in a recirculating aquaculture system. The fish from the Fritz Lipmann Institute (FLI) facility are under §11 license no. J-0003798 and §4 license O_DV_21_24. For the fish raised at the Max Planck Institute for Biology of Ageing (MPI-AGE), the holding license (§11TSchG) was 576.1.36.6.G12/18; for the preparation of tissues (§4 TSchG) the license number was MPIa_Anzeige§4_RB.16.005.
To collect the fish plasma, all the fish received lethal anesthesia with 1.5 g l−1 Tricaine mesylate solution; then, an incision with a scalpel at the level of the caudal peduncle was made. After amputation, the amputated portion was immersed into a 50-μl drop of plasma collection buffer (dPBS1X + 1:500 diluted solution containing gentamicin 5 mg ml−1 and amphotericin B 125 μg ml−1) inside a Petri dish. After this procedure, we collected blood using a pipette into a 1.5-ml Eppendorf tube. The collected blood samples were centrifuged at 1,500g and 4 °C for 20 min. The supernatant was collected into a new 1.5-ml Eppendorf tube. Next, the sample was immediately stored at −80 °C.
The sequencing data associated with this study are available under the project name ‘Antibody Repertoire Sequencing Reveals Systemic and Mucosal Immunosenescence in the Short-lived Turquoise Killifish’.
To isolate immune cells from the kidney marrow, both kidneys were collected in a 1.5-ml Eppendorf tube with 900 μl of dPBS1X. To obtain a single cell suspension, the tissue was dissociated by gently pipetting inside the Eppendorf tube until the solution was homogeneous. Then, the sample solution was filtered through a 35-μm nylon membrane in the cap of a 5-ml fluorescence-activated cell sorting tube and immediately transferred into a new 1.5-ml Eppendorf tube. The sample was centrifuged at 400g and room temperature for 4–5 min. Next, the supernatant was discarded and the pellet was resuspended in 100 μl dPBS1X. Then, 900 μl of milliQ water were added to the blood sample. The sample was manually inverted for 20 s, then 100 μl PBS10X were added to the sample, which was centrifuged at 400g and room temperature for 4–5 min. Finally, the supernatant was discarded and the cell pellet was resuspended once in dPBS1X (the cell number was quantified before the washing passage). For immune cells committed to scRNA-seq, we further separated immune cells from cell debris using a BD FACSAria III Cell Sorter. After resuspension in dPBS1X, cells were centrifuged at 500g at 4 °C for 10 min, the supernatant was completely discarded and the remaining pellet inside the 1.5-ml Eppendorf tube was stored in liquid nitrogen (and afterward at −80 °C). We used the same set of adult and aged-adult male individuals to perform all the proteomics analyses.
scRNA-seq library preparation
To prepare the libraries for 10X scRNA-seq, we used the 10X Genomics Chromium Single Cell 3’ Library (PN-120234) & Gel Bead Kit v2 (PN-120235) according to the manufacturer’s instructions. We isolated immune cells from the kidney marrows of three young-adult (8-week-old) and three geriatric (21-week-old) fish. Then, for each sample, single-cell suspensions were encapsulated into gel beads in emulsion (GEMs) using the Chromium Controller. Barcoded reverse transcription primers were used to capture the polyadenylated mRNA; complementary DNA (cDNA) synthesis was performed within the GEMs. After GEM incubation, GEMs were broken and cDNA was purified. The cDNA underwent amplification and library construction through end repair, A-tailing, adapter ligation and sample index PCR. cDNA yield and size distribution was measured using an Agilent Bioanalyzer. Sequencing of the single-cell libraries was performed by the Cologne Centre on Genomics, Cologne, Germany.
Protein isolation and TMT labeling
Proteins from immune cells isolated from frozen kidney marrows were extracted with a guanidine chloride protocol; 20 μl of lysis buffer (6 M guanidinium chloride, 2.5 mM Tris(2-carboxyethyl) phosphine, 10 mM chloroacetamide, 100 mM Tris-HCl) were added to the pellet. The samples were heated for 10 min at 95 °C and then lysed with a Bioruptor for ten cycles of 30-s sonication and 30-s break on high performance. The samples were then centrifuged at 20,000g for 20 min. The supernatant was transferred into a new tube and the protein concentration was measured using NanoDrop. To prepare peptides from both serum and immune cell protein samples, 300 μg of protein per sample was digested with 1:200 trypsin (w/w) at 37 °C overnight. The digest was then acidified with formic acid (FA) to a final concentration of 1% to stop tryptic digest. The samples were then centrifuged at 20,000g for 10 min to pellet any remaining debris. The peptides were then cleaned with a StageTip protocol (C18-SD tips), including a series of wetting, equilibrating, washing and eluting steps. The C18-SD tips were first washed with 200 μl methanol using centrifugation for 1 min, followed by a wash with 200 μl 40% acetonitrile/0.1% FA using centrifugation for 1 min. The tips were then equilibrated with 200 μl 0.1% FA using centrifugation for 1 min. The digests were then loaded onto the tips and centrifuged for 2 min to ensure proper loading. The tips were washed twice with 200 μl 0.1% FA, followed by elution of the peptides with 100 μl 40% acetonitrile/0.1% FA by centrifuging for 4 min at 1,500g. The eluates were dried in a Speed-Vac at 45 °C for 45 min, then resuspended in 0.1% FA and quantified with NanoDrop; 4 μg of the eluted peptides were dried and reconstituted in 9 μl of 0.1 M tetraethylammonium bromide. Labeling with TMTs (TMTproTM 16plex, Thermo Fisher Scientific) was carried out according to the manufacturer’s instructions.
Proteomics data preprocessing
Proteomics data were analyzed using MaxQuant v.1.6.10.43 (ref. 80). Peptide fragmentation spectra were searched against the canonical sequences of the N. furzeri proteome (downloaded September 2019 from UniProt). Protein names and primary gene names, corresponding to the UniProt IDs were downloaded from UniProt and used to annotate the data. Methionine oxidation and protein N-terminal acetylation were set as variable modifications; cysteine carbamidomethylation was set as a fixed modification. The digestion parameters were set to ‘specific’ and ‘Trypsin/P’. Quantification was set to ‘Reporter ion MS3’. The isotope purity correction factors, provided by the manufacturer, were imported and included in the analysis. The minimum number of peptides and razor peptides for protein identification was one; the minimum number of unique peptides was zero. Protein identification was performed at peptide spectrum matches and a protein false discovery rate of 0.01. The ‘second peptide’ option was on. TMT reporter intensities were normalized using vsn81 and log2-transformed in R v.3.4.3 (ref. 82). We calculated the differences between log2 median protein abundance levels of adult and aged-adult samples to determine differential expression. Statistical significance was assigned based on Wilcoxon signed-rank test using the ‘wilcox.test’ function in R.
GSEA on proteomics data
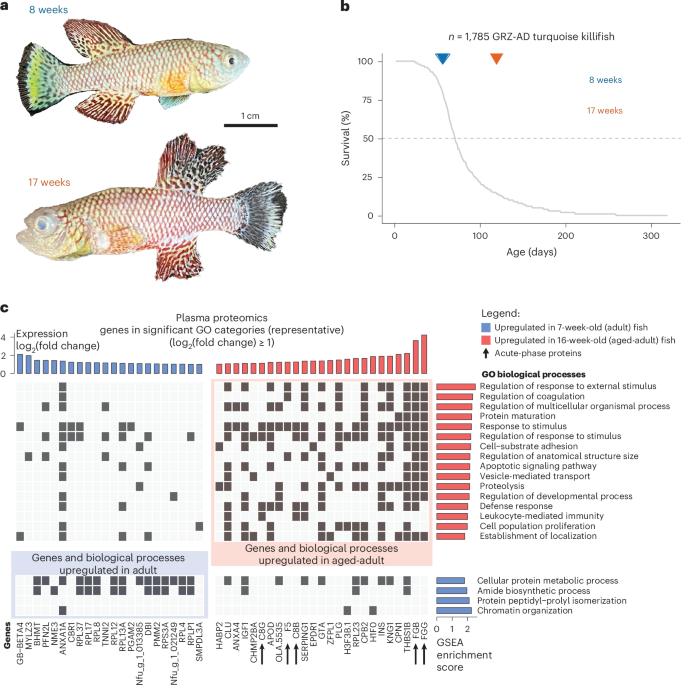
GSEA was performed using the ‘gseGO’ function in the ‘clusterProfiler’ package (v.4.2.2)83, using human gene symbols and the ‘org.Hs.eg.db’ annotation package in R. The Ensembl database and R ‘biomaRt’ package (v.2.50.2) were used to map ortholog genes between human and N. furzeri genes (date of access: 17 February 2022). We analyzed the GO Biological Processes categories with a minimum of ten and maximum of 500 annotated genes. We used Benjamini–Yekutieli correction for multiple testing and considered a Benjamini–Yekutieli-corrected P < 0.05 as significant. Details of human-*N. furzeri* orthology mapping are explained under ‘Helper functions/data -> Gene ID & Orthology mapping’. Two outputs from the ‘gseGO’ function were used: core enrichment genes (that is, genes that contribute most to the enrichment result) and normalized enrichment score. As the GO enrichment results gave many significant GO categories but most of them showed overlaps, we used an in-house method to choose representative GO categories for visualization and summarization purposes. To choose representative GO categories, we calculated the Jaccard similarity between GO categories based on core enrichment genes. We then performed hierarchical clustering of the similarity matrix, cutting the tree at different levels (20–70 clusters). We calculated the median Jaccard index within each cluster. We took the minimum number of clusters, k, where at least half of the clusters had a median Jaccard index of 0.5 or higher. This resulted in 20 clusters (that is, representatives) for plasma proteomics and 50 clusters (that is, representatives) for kidney marrow proteomics.
scRNA-seq dataset analysis
N. furzeri primary genome assembly and genome annotations were downloaded from Ensembl (v.105). Genome annotations were filtered to include only the protein-coding immunoglobin or T cell receptor genes using the ‘cellranger mkgtf’ command. A custom reference for N. furzeri was built using the ‘cellranger mkref’ command. A count matrix was generated for each sample in each run separately using the ‘cellranger count’ function. Only cells with a minimum of 200 and a maximum of 2,500 features were kept. First, all samples in each run were log-normalized independently using the ‘NormalizeData’ function in Seurat. Next, 2,000 variable features were selected using the ‘vst’ method implemented in the ‘FindVariableFeatures’ function in Seurat. For clustering, we used shared nearest neighbor graph construction with a resolution of 0.3, which provided the last most stable clustering based on the clustering tree. Then, differentially expressed markers for each cluster were identified. All genes expressed in at least 25% of cells in a cluster were tested. Based on the resulting marker list, cell types were hand-annotated using CellMarker 2.0 (http://bio-bigdata.hrbmu.edu.cn/CellMarker/) as the reference database. As four clusters suggested contamination from gonads, we excluded those cells from the analysis. The same steps were repeated except we used the first 20 principal components for clustering, uniform manifold approximation and projection, and t-SNE plots. The previous steps for cell clustering and cell type annotation were repeated, yielding 16 clusters. To further refine these annotations, we reanalyzed our dataset using a recent scRNA-seq dataset30. First, to assess the accuracy of reference-based annotation, we subsampled 20% of kidney cells from male fish (436 cells) from the Teefy et al.30 dataset and reannotated these cells using Seurat (v.3.2.2) in R, with the remaining cells from their dataset as the reference. This approach achieved approximately 80% accuracy across clusters, although progenitor cells showed lower annotation performance, with accuracy around 50–60%. Subsequently, we applied the Teefy et al.30 dataset as a reference to reannotate our own data. Most clusters aligned well with our manual annotations, with over 60% of cells in each cluster annotated consistently. Exceptions included clusters 4 and 13, which corresponded closely to multiple cell types without a clear, distinct annotation. We decided to maintain the labeling of clusters 4 and 13 as B cells and NK cells, respectively; a different labeling was suggested using the Teefy et al.30 annotation. In particular, cluster 4 was suggested to contain both B cells and erythrocytes using the Teefy et al.30 annotation; however, it shows cluster-specific expression of B cell markers like CD74b and CD79a, not usually expressed by erythrocytes84,85. Cluster 13 was suggested to contain macrophages and erythrocytes by the Teefy et al.30 annotation. However, we decided to maintain NK cell labeling for this cluster because it is very close to other lymphocyte clusters and expresses both macrophages and lymphoid markers.
Proteomics GO category representation in scRNA-seq clusters
Four GO representative categories—Cellular detoxification, DNA repair, DNA replication and Immune cell activation—were chosen for further investigation in the scRNA-seq data based on the results of the proteomics dataset from the kidney marrow. These GO categories represent 35, 14, 55 and 15 GO categories, respectively. All human genes associated with these categories were obtained by propagating gene membership in the GO hierarchical tree using the ‘GO.db’ (v.3.14), ‘AnnotationDbi’ (v.1.56.2) and ‘org.Hs.eg.db’ (v.3.14.0) packages in R (date of access: 7 April 2022). We then mapped this information to the orthologs in N. furzeri, intersecting with the scRNA-seq dataset, getting the genes with an absolute value of log2(fold change) of 1 in the kidney marrow proteomics data and only using the genes present in only one of the four categories. For each cell in the dataset, we calculated the percentage of the expressed genes in each representative category and divided this number by the percentage of expressed genes among all genes detected using the scRNA-seq. Thus, we aimed to normalize the overall transcriptional profile of cells and obtained an OR showing the enrichment of genes specific to the GO categories of interest. We colored cells on t-SNE with this OR.
Induction of DNA damage via UV irradiation
To induce DNA damage, primary kidney marrow cells from a young-adult male fish were exposed to 100 J m2 using a standard UV crosslinker (cat. no. BLX312 UV, Vilber Lourmat). UV-irradiated cells were stained for immunofluorescence with two different γH2AX antibodies.
Samples preparation for FISH
Immune cells freshly isolated from kidney marrow were fixed 30 min in 4% paraformaldehyde (PFA) and washed once in dPBS1X. Then, cells were resuspended in 300 μl hybridization mixture (formamide 70%, blocking reagent 1×, Tris-HCl, pH 7.4, 10 mM, locked nucleic acid probe 0.5 mM). Samples were kept for 5 min at 80 °C and left at room temperature for 2 h. For imaging, samples were washed twice in dPBS1X and acquired with an Amnis ImageStreamX MkII Imaging Flow Cytometer using ×60 magnification and ex/em wavelength of 642/690 nm to detect the locked nucleic acid signal.
ImageStream sample preparation and processing
Immune cells freshly isolated from the kidney marrow were fixed for 30 min in 4% PFA, then centrifuged at 400g for 10 min and washed once in dPBS1X. Next, the pellet was resuspended in 500 μl permeabilization buffer (dPBS1X + 0.1% Triton X-100) and incubated for 15 min. Samples were centrifuged at 400g for 10 min; then, the cell pellet was resuspended in 500 μl of blocking buffer (permeabilization buffer + 2% BSA) and incubated 60 min at room temperature. The samples were centrifuged at 400g for 10 min, the cell pellet was resuspended in 90 μl of primary antibody solution (blocking buffer + 1:200 Rabbit anti-γH2AX antibody (Ser139) (20E3) Rabbit monoclonal antibody, cat. no. 9718S, Cell Signaling Technology) and samples were incubated overnight at 4 °C. Then cells were washed once in dPBS1X, the pellet was resuspended in 90 μl secondary antibody solution (blocking buffer + 1:500 Goat anti-rabbit IgG Alexa Fluor 647 antibody cat. no. A27040, Invitrogen) and the samples were incubated at room temperature for 45 min in the dark. Finally, cells were washed twice in dPBS1X and the cell pellet was resuspended in 40 μl of DAPI staining solution (dPBS1X + DAPI 1 μg ml−1). Immune cells not treated with primary antibody solution and DAPI staining solution were used as the negative control. All the images were acquired with an Amnis ImageStreamX MkII Imaging Flow Cytometer using ×60 magnification and an ex/em wavelength of 405/468 nm to detect the DAPI signal and 642/690 nm to detect the γH2AX signal.
Giemsa staining of immune cells
Immune cells freshly isolated from kidney marrow were sorted into myelomonocyte, progenitor and lymphocyte populations based on their FSC and SSC properties using a BD FACS Aria III cell sorter. Immune cells from each sorted population were fixed for 30 min in 4% PFA, washed once in dPBS1X and seeded on a glass slide using Cytospin 4 centrifuge. Afterwards, slides were immersed in Giemsa stain solution for 40 s at room temperature. After staining, cells were washed gently three times with distilled water to remove excess stain; then, each sample was imaged at ×40 magnification with an EVOS XL Core Imaging System.
Classification of cell type using ImageStream and machine learning
We analyzed all the raw data (.rif files) using Amnis IDEAS v.6. First, to train the machine learning algorithm (linear discriminant analysis machine learning algorithm), we created a merged.rif file containing the raw data of all the adult and aged-adult samples; then, we manually labeled all cell types based on specific cytometry features86. The training was performed by using 316 cells manually annotated as ‘erythrocytes’, 291 cells manually annotated as ‘progenitor-1’, 137 cells manually annotated as ‘myelomonocytes’, 235 cells manually annotated as ‘lymphocytes’, 261 cells manually annotated as ‘polynucleated cells’ and 116 cells manually annotated as ‘progenitor-2’. To differentiate between the two classes of progenitors, based on the available fish literature, we used the following approach: progenitor-1 cells have an irregular cell shape, no black dots in the cytoplasm and low cell brightness in the brightfield. Instead, progenitor-2 cells have a round shape, no black dots in the cytoplasm and a darker color under brightfield. In addition, both cell types are smaller than myeloid cells and are larger than lymphoid cells. To test the accuracy of the model, we manually labeled randomly for each training class additional cells as ‘TRUE’ representatives of the class (corresponding to 19% of the training class cell number). These additional ‘TRUE’ classes do not overlap with the cells used in classes for the training algorithm. Then, for each ‘TRUE’ class, we checked the ratio of predicted classes. A classification index of zero was used as a threshold to determine the predicted classes. The percentages of the true cell populations in predicted classes are provided as a measure of accuracy. Then, we used the initial training set to classify different clusters predicted in each single.rif data file for adult and aged-adult samples and in the two merged.rif files we generated for both adult and aged-adult conditions.
γH2AX/telomere costaining and microscopy
Immune cells freshly isolated from kidney marrow were fixed for 30 min in 4% PFA and processed as explained in the previous section (‘ImageStream sample preparation and processing’). After sample incubation with secondary antibody, immune cells were refixed in 4% PFA + 0.1% Triton X-100 at room temperature for 10 min. Then, the sample was centrifuged at 400g for 10 min; the pellet was resuspended in 100 μl of 10 mM glycine diluted in double-distilled H2O, followed by a 30-min incubation at room temperature. Cells were washed once in dPBS1X and resuspended in 300 μl hybridization mixture (formamide 70%, blocking reagent 1×, Tris-HCl, pH7.4, 10 mM, telomeric PNA probe 0.5 mM). Then, the samples were kept for 5 min at 80 °C and left at room temperature for 2 h. For imaging, the samples were washed twice in dPBS1X and dropped on a slide (adding DAPI). All images were acquired using PALM laser-capture microdissection and an optical tweezer microscope TCS SP8 STED-3× using a magnification of ×100 and an ex/em wavelength of 405/468 nm to detect the DAPI signal and 642/690 nm to detect the γH2AX signal.
CFU immune cells cluster assay
Immune cells freshly isolated from the kidney marrow of young-adult (8-week-old) and aged (17-week-old) fish were resuspended in Roswell Park Memorial Institute (RPMI) 10% medium (RPMI + HEPES 1:40 + heat-inactivated FCS 10% + 1:500 diluted solution containing gentamicin 5 mg ml−1 and amphotericin B 125 μg ml−1) and seeded at a concentration of 200,000 cells per well in the wells of a 96-well plate. These cells were then treated or not treated with different concentrations of LPS (cat. no. L6529, Sigma-Aldrich) and incubated at 28 °C for 24 h. Then, each well of the plate was imaged using an EVOS XL Core Imaging System (AMEX1000, Thermo Fisher Scientific) at ×40 magnification. To check the fisetin effect on killifish immune cells, cells seeded once on the 96-well plate were preincubated in the presence of 15 μM fisetin at 28 °C for 24 h before medium change and LPS treatment, as explained above. Quantification of the clusters in each well was made blindly using Fiji; the number of the clusters in the wells stimulated with LPS was calculated by subtracting the number of clusters obtained in the negative control from those present in each experimental condition.
β-Gal staining
Immune cells freshly isolated from the kidney marrow of young-adult (8-week-old) and aged (17-week-old) fish were fixed for 30 min in 2% PFA, then centrifuged at 400g for 10 min and washed once in dPBS1X. Afterwards, samples were stained according to the manufacturer’s instructions for the CellEvent Senescence Green Flow Cytometry Assay Kit. All images were then acquired using the Amnis ImageStreamX MkII Imaging Flow Cytometer using ×60 magnification and an ex/em wavelength of 488/530 nm to detect the β–gal signal.
Bayesian hierarchical model of β-gal in immune cells from young and aged killifish
To assess the changes in β-gal staining between aged and nonaged adult killifish, we used a hierarchical Bayesian model, using the rstan package in the R programming environment. The model was designed to assess the effects of age and cell type on the expression of β-gal (bgal_cl) across different immune cell types.
Data preparation
The dataset included measurements of β-gal levels (bgal_cl) in five distinct immune cell types—erythrocytes (E), lymphocytes (L), monocytes (M), progenitor-1 (P1) and progenitors-2 (P2)—collected from killifish at two different ages (8 weeks and 17 weeks). The variable week was transformed into a binary variable (age_binary), where 0 indicated young killifish (8 weeks) and 1 indicated aged killifish (17 weeks). The cell_type variable was converted into a numeric factor to facilitate its use in the hierarchical model.
Model specification
Given the nature of the β-gal measurements, which are constrained between 0 and 1, a Beta distribution was chosen as the likelihood function for this hierarchical Bayesian model. The Beta distribution is particularly suited for modeling proportions and rates, making it a better fit than the Gaussian distribution for these data.
Parameters
-
alpha: Overall intercept representing the baseline level of β-gal expression across all cell types and ages.
-
beta_age: Coefficient representing the main effect of age on β-gal expression.
-
alpha_cell: A vector of cell-type-specific intercepts, capturing the baseline level of expression for each cell type.
-
beta_age_cell: A vector of coefficients representing the interaction effect between age and each cell type, allowing the effect of age to vary according to cell type.
-
phi: Precision parameter of the Beta distribution, which controls the variance independently of the mean.
Priors
-
Non-informative normal priors were placed on the intercept (alpha), the main effect of age (beta_age), the cell-type-specific intercepts (alpha_cell) and the interaction effects (beta_age_cell), with a mean of 0 and an s.d. of 10.
-
A weakly informative Gamma prior was used for the precision parameter (phi), ensuring that the model can accommodate a wide range of variances in β-gal expression levels.
Likelihood
The likelihood function assumed that the observed β-gal levels (bgal_cl) followed a Beta distribution, with a mean parameterized as a linear combination of the overall intercept, the main effect of age, the cell-type-specific intercept and the interaction between age and cell type, as follows:
bgal_cl[i] ~ Beta(mu[i] * phi, (1 – mu[i]) * phi)
where mu[i] = logit^(-1)(alpha + beta_age * age_binary[i] + alpha_cell[cell_type[i]] + beta_age_cell[cell_type[i]] * age_binary[i]).
Model fitting and diagnostics
The model was fitted using the stan() function, with 2,000 iterations across four Markov Chain Monte Carlo chains. Convergence was assessed using trace plots and the Gelman–Rubin diagnostic, with no indication of non-convergence. We further compared the performance of the likelihood function based on a Beta distribution to one using a normal distribution. We performed model cross-validation using leave-one-out in rstan. The analysis is available on our GitHub page at https://github.com/valenzano-lab/public/tree/main/GM.
Statistics and reproducibility
All experiments used turquoise killifish (N. furzeri, GRZ strain) raised under standardized husbandry conditions in recirculating aquaculture systems at MPI-AGE and FLI. Age groups and sample sizes were chosen based on prior experience with this model, feasibility of tissue and cell yields for each assay, and established practice for comparable omics and cytometry experiments in killifish. No statistical method was used to predetermine sample size. Biological replicates (individual fish) are stated in the figure legends, and include (non-exhaustive): plasma TMT proteomics (n = 5 adult versus n = 5 aged-adult male fish); kidney marrow TMT proteomics (n = 5 adult versus n = 5 aged-adult male fish, same individuals as for the plasma proteomics); ImageStream cytometry for cellularity, proliferation and γH2AX (n = 5 adult versus n = 4 aged-adult male fish); histology (Sirius Red/Fast Green, n = 3 adult versus n = 3 aged-adult); and LPS aggregate formation assays (typically n = 3 adult versus n = 3 aged-adult). Technical replication was used where appropriate (for example, repeated antibody validation for γH2AX after UV irradiation; repeated FISH validation experiments; multiple images and fields quantified per individual for histological readouts as described).
Data exclusion
For scRNA-seq, one of six sequenced samples did not meet the quality and read-depth criteria and was excluded before downstream analysis; the remaining five samples were retained. In the scRNA-seq analysis, cells mapping to clusters indicative of gonadal contamination were excluded before final clustering and annotation; downstream analyses were repeated on the filtered dataset. Apart from these predefined quality control and contamination-filtering steps, no data were excluded from the analyses.
Randomization and blinding
Allocation of animals to age groups was determined according to chronological age; therefore, it was not randomized. The experiments were not randomized. Investigators were generally aware of age group allocation during the experiments and outcome assessment; however, for the colony-forming unit/aggregate quantification, cluster counts were performed blinded in Fiji. Overall, the investigators were not blinded to allocation during the experiments and outcome assessment.
Statistical analyses
Statistical analyses were performed in R v.3.6.1 unless otherwise indicated. For two-group comparisons of continuous outcomes with small sample sizes and non-normal distributions, two-sided nonparametric Wilcoxon signed-rank tests were used (for example, cytometry-derived proportions and intensities; proteomics differential abundance testing via wilcox.test). For the proteomics GSEAs, multiple-testing correction was performed using Benjamini–Yekutieli adjustment, with a Benjamini–Yekutieli-adjusted P < 0.05 considered significant; reporter ion intensities were variance-stabilized (vsn) and log2-transformed before testing. Where Bayesian modeling was used (β-gal), a hierarchical Beta regression model was implemented in R using rstan, with four Markov Chain Monte Carlo chains and 2,000 iterations per chain; model convergence was assessed using trace plots and Gelman–Rubin diagnostics; model comparison and cross-validation was performed using leave-one-out procedures as described. Exact statistical tests, sidedness, multiple-testing procedures, thresholds for significance and the definition of box plot elements (median, IQR, whiskers) are reported in the figure legends. The code used for the omics analyses and the KIAMO Shiny app is publicly available (the GitHub links have been provided in the paper), and the primary datasets are deposited in public repositories as stated under ‘Data availability’.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.