
Mice and reagents
Animal studies (EC2020-005, EC2021-048, EC2022-004 and EC2023-039) were approved by the ethical committee of Ghent University Center for Inflammation Research, Belgium. Embryonic C57BL/6J mouse work was performed in accordance with the Institutional Animal Care and Use Committee and relevant guidelines of Boston Children’s Hospital and Masaryk University. C57BL/6J mice (Mus musculus), B6.129S4-Pdgfratm11(EGFP)Sor/J49 and B6(C)-Ccr2tm1.1Cln/J (strain 027619)50 were maintained in the VIB Center for Inflammation Research animal facility. Mice were housed in individually ventilated cages in specific pathogen-free conditions, under a 14–10 h light–dark cycle, at 20–22 °C and 40–60% relative humidity, with food and water ad libitum. We used a mix of male and female mice of 7–12 weeks old, unless specified elsewhere. For studies on aged mice, we compared 22- and 82-week-old male C57BL/6J mice. No statistical methods were used to predetermine sample sizes but our sample sizes are similar to those reported in previous publications. For the induction of systemic inflammation, we used male and female C57BL/6J mice that received i.p. injection of 300 µg 200 µl−120 g−1 body weight (BW) LPS (Sigma, L-5886). For in vivo blood vessel tracing, we used male and female 7–12-week-old C57BL/6J mice that received i.v. injection of 600 µg 200 µl−120 g−1 BW concanavalin A-FITC (Vector Labs, VEC.FL-1001) diluted in PBS solution. For in vivo tracer leakage, we used naive and systemically inflamed male and female 7–12-week-old C57BL/6J mice that received either i.v. or i.c.v. injection of dextran-3000 or 10,000 MW-biotin-lysin (Thermo Fisher, D7135). The i.v. dose was 2.5 mg 200 µl− 120 g−1 BW. For i.c.v. injections, mice were first anesthetized with isoflurane and mounted on a stereotactic frame. The body temperature of the mice was maintained at 37 °C using a heating pad. A volume of 5 μl of 100 mg ml−1 was injected at a rate of 1 µl min−1 into the left lateral cerebral ventricle with a Hamilton needle, for which the injection coordinates were measured from the bregma (anteroposterior 0.07 cm, mediolateral 0.1 cm and dorsoventral 0.2 cm) using the Franklin and Paxinos Mouse Brain Atlas. Embryonic Tbx18tm3.1(cre/ERT2)Sev/J (ref. 20) × Ai14fl/fl (Gt(ROSA)26Sortm14(CAG-tdTomato)Hze) mice and Mesp1tm(cre)Ysa (ref. 18) × Ai14fl/fl (Gt(ROSA)26Sortm14(CAG-tdTomato)Hze) experiments were performed in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care and were handled in accordance with protocols approved by the University of Colorado Anschutz Medical Campus Institutional Animal Care and Use Committee. Breeding, mice set-ups and tamoxifen administration were performed as described previously21. For all experimental set-ups, mice were randomly assigned to predetermined sized groups. Mice were allocated sequential numbers to keep experimenters blind during tissue extraction/processing/analyses. No animals or data points were excluded from analyses for any biological reasons; however, due to the difficulty in getting good-quality sections/samples of the ChP base, many mice/samples did not end up being useful.
Human samples
Human samples were obtained under an Institutional Review Board (IRB)-approved protocol at Boston Children’s Hospital. A neuropathologist (H.G.W.L.) reviewed the human tissue specimens and recorded non-identifying information. Human brain tissue samples were obtained from autopsies performed at Boston Children’s Hospital as part of the requested clinical service. The example shown (5-week-old female, congenital diaphragmatic defect hepatomegaly) did not have identified neurological disease although the gyral pattern was described as nonspecifically abnormal. Parents provided signed informed consent for research based on a written explanation of the anticipated use of samples not required for clinical purposes. This study was approved by Institutional Postmortem Research Committee of Boston Children’s Hospital (IRB-P00003747). Use is compliant with Health Insurance Portability and Accountability Act informed consent procedures.
Tissue processing and immunostainings
Mice were sedated with a mix of ketamine (20 mg ml−1) and xylazine (4 mg ml−1), followed by transcardial perfusion with PBS with 0.2% heparin, 5,000 IU ml−1, Wockhardt. Brains were dissected and fixed in 4% PFA at 4 °C for 1–4 h, followed by a 10–20–30% sucrose gradient, embedding in Neg50-OCT cryogel and storage at −70 °C. Cryosections of 12–18-µm thick were cut using the Cryostar NX70. Sections were then post-fixed with 4% PFA for 10 min. For CDH1/ZO1/LAMININ staining, sections underwent antigen retrieval in a cooker for 5 min with 1× citrate based unmasking solution (pH 6; H3300 Vector), followed by cooling down at 4 °C for 30 min. After post-fix or antigen retrieval, sections were washed with PBS and blocked for 1 h at room temperature with PBS containing 0.1% Triton X-100, 0.5% BSA and 2% serum. Serum was matched to the species of secondary antibodies. The same blocking buffer was used to dilute primary and secondary antibodies. Primary antibodies were incubated overnight at 4 °C, followed by washes in PBS and fluo-conjugated secondary antibody incubation for 2 h at room temperature. Secondary antibodies used in this study were goat anti-mouse IgG (H + L) cross-adsorbed secondary antibody, DyLight 488 (1:500 dilution; Thermo Fisher Scientific, 35503), goat anti-rabbit IgG (H + L) cross-adsorbed secondary antibody, DyLight 488 (1:500 dilution; Thermo Fisher Scientific, 35553), goat anti-rabbit IgG (H + L) highly cross-adsorbed secondary antibody, Alexa Fluor 568 (1:500 dilution, Thermo Fisher Scientific, A11036), goat anti-rat IgG (H + L) cross-adsorbed secondary antibody, Alexa Fluor 633 (1:500 dilution; Thermo Fisher Scientific, A21094), goat anti-rabbit IgG (H + L) cross-adsorbed secondary antibody, DyLight 633 (1:500 dilution; Thermo Fisher Scientific, 35563), streptavidin protein, DyLight 633 (1:500 dilution; Thermo Fisher Scientific, 21844), goat anti-chicken IgY (H + L) secondary antibody, Alexa Fluor 488 (1:500 dilution; Thermo Fisher Scientific, A11039) and goat anti-rabbit IgG (H + L) cross-adsorbed secondary antibody, Alexa Fluor 594 (1:500 dilution; Thermo Fisher Scientific, A11012). Primary antibodies used in this study were rabbit anti-CLDN11 (1:800 dilution; Thermo Fisher Scientific, 36-4500); rat anti-CD31 (1:100 dilution; BD Pharmigen, 553370); mouse anti-ACTA2-Cy3 (1:400 dilution; Sigma Aldrich, C6198); rabbit anti-ZO1 (1:500 dilution; Invitrogen, 617300); mouse anti-CDH1 (1:200 dilution; Becton Dickinson, 610181); chicken anti-GFP (1:750 dilution; Abcam, ab13970) and rabbit anti-LAMININ (1:500 dilution; Thermo Fisher Scientific, PA1-16730). After additional washing in PBS and incubation with 4,6-diamidino-2-phenylindole for 30 min at room temperature, sections were mounted using PVA-DABCO mounting medium. For the visualization of i.v. injected dextran-3000 or 10,000 MW-biotin-lysin (Thermo Fisher Scientific, D7135), sections were incubated with Streptavidin-DyLight633 (1:1,000 dilution; Thermo Fisher Scientific, 21844) diluted in PBS for 30 min at room temperature. Slides were imaged with a Zeiss LSM780 confocal microscope (Carl Zeiss) and analyzed using ImageJ. For colocalization analyses, we used QuPath to calculate the intersect and finally the %CLDN11 signal overlapping with either 3 kDa dextran signal or lineage-tracing AI14 reporter signal. Data shown are % colocalization averaged per mouse, with a minimum of two sections imaged per ventricle per mouse. For quantification of CCR2+ cells per volume and distances to CLDN11+ base and for ZO1/CDH1 mean and integrated density analyses we used QuPath 0.4.0 software.
RNA ISH experiments
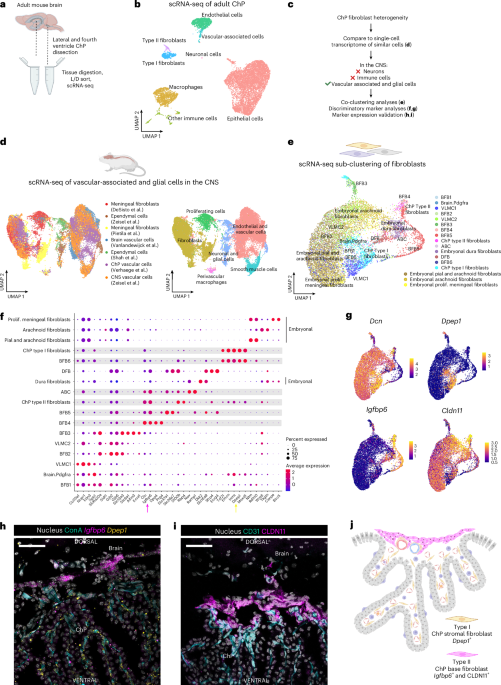
For the analyses of Cldn11 and Igfbp6 expression in the developing mouse brain, we investigated the Developing Mouse Brain dataset from the Allen Brain Atlas (developingmouse.brain-map.org/). For the analyses of Igfbp6 and Dpep1 expression in adult mouse brains, we performed multiplex RNA-FISH experiments using Molecular Instruments technology. Tissues were collected as described above, with the sole exception of 24 h fixation in 4% PFA at 4 °C. In brief, cryosections were air dried at room temperature for 15 min, washed in PBS and then baked at 55 °C for 30 min. After additional washing, we incubated Proteinase K 5 µg ml−1 for 3 min at room temperature, followed by washing and 15 min post-fix in 4% PFA. From this point on, we performed steps as recommended by the company and online available protocol HCR RNA-FISH protocol for fresh frozen or fixed frozen tissue sections. We used Dpep1-B4 (NM_007876.2) and Igfbp6-B2 (NM_008344.3) probes. For the analyses of IGFBP6 and CLDN11 in human postmortem formalin-fixed paraffin-embedded samples, we performed RNAscope analyses. Formalin-fixed paraffin-embedded tissue sections were stained with hematoxylin and eosin according to standard methods to identify sections with ChP. Consecutive sections from positively identified sections were used for RNAscope and immunolabeling. In brief, IGFBP6 mRNA was localized using the RNAscope probe (ACD Bio, cat no. 496061) and the RNAscope Multiple Fluorescent v2 Assay (cat. no. 496069). Immediately following the completion of the assay, CLDN11 immunostaining was performed using rabbit anti-Cldn11 antibody (1:500 dilution, Thermo, cat no. 36-4500). In brief, tissue sections were blocked for 1 h with 1× PBS + 5% goat serum, followed by incubation with primary antibody at 4 ˚ C overnight and then with Alexa Fluor secondary antibodies (1:500 dilution, goat anti-rabbit 594, Thermo Fisher A11012) and nuclei were counterstained with Hoechst. Slides were imaged with a Zeiss LSM780 confocal microscope (Carl Zeiss) and analyzed using ImageJ (1.52p).
Herovici staining
For the analyses of collagens in the ChP, Herovici staining was performed. In brief, mice were perfused with PBS heparin and brains were isolated and fixed in 4% PFA overnight at 4 °C. Samples were dehydrated through an ethanol gradient, xylene and embedded in paraffin. Then, 5-µm thick sections were cut using the HM340E microtome and dried overnight. Samples were rehydrated over xylene and ethanol steps and stained for 10 min with the acid-resistant nuclear staining Weigert’s iron hematoxylin solution (Sigma). After rinsing with tap water for about 5 min, samples were incubated with the staining solution (41% picric acid, 4.5% acid fuchsin, 45% methyl blue, 9% glycerol and 0.45% lithium carbonate) for 2 min and washed with 1% acetic acid for 2 min. Finally, samples were again dehydrated with ethanol, cleared with xylene and mounted using Entellan (Merck Millipore). All steps were performed at room temperature. Slides were imaged with Zeiss AxioScan microscope.
RNA extraction and RT–qPCR analysis
ChP tissues were dissected as described previously51. ChP from perfused mice were snap-frozen in liquid nitrogen and stored at −80 °C. TRIzol (Invitrogen) was used to homogenize the tissue with the TissueLyser (QIAGEN) in a tube with zirconium oxide beads. Afterwards, chloroform was added and the homogenate was separated in phases by centrifugation at 20,000g for 15 min at 4 °C. The upper phase was collected and RNA extracted using the Aurum total RNA kit (Bio-Rad) according to the manufacturer’s guidelines. RNA concentration was measured using the Nanodrop-1000 (Thermo Scientific) and reverse-transcribed with the SensiFAST cDNA Synthesis kit (Bioline) to generate cDNA. The primers used were Cldn11 Fw ATGGTAGCCACTTGCCTTCAG; Cldn11 Rev AGTTCGTCCATTTTTCGGCAG; IL6 Fw TAGTCCTTCCTACCCCAATTTCC; IL6 Rev TTGGTCCTTAGCCACTCCTTC; Rpl Fw CCTGCTGCTCTCAAGGTT; Rpl Rev TGGTTGTCACTGCCTCGTACTT; Ubc Fw AGGTCAAACAGGAAGACAGACGTA; Ubc Rev TCACACCCAAGAACAAGCACA; Gapdh Fw TGAAGCAGGCATCTGAGGG; Gapdh Rev CGAAGGTGGAAGAGTGGGAG; Hprt Fw AGTGTTGGATACAGGCCAGAC; Hprt Rev CGTGATTCAAATCCCTGAAGT; S100b Fw TACTCGGACACTGAAGCCAG; S100b Rev CCCGGAGTACTGGTGGAAGA; Rbp4 Fw AAGGGTCATATGAGCGCCAC; Rbp4 Rev CGTGTCGATGATCCAGTGGT; Igfbp6 Fw GCTGCTAATGCTGTTGTTCGC; and Igfbp6 Rev GCACTTAGGGCTGTAGACCC. A qPCR reaction was performed using the SensiFAST SYBR No-ROX kit (Bioline) on the Roche LightCycler 480 (Applied Biosystems). Data were analyzed using the qbase+ software. Values shown in figures are relative expression values normalized to geometric means of the reference genes.
Tissue processing and scRNA-seq
Mice for all groups were first sedated as described above, transcardially perfused with PBS with 1% heparin after which brain tissue was excised and lateral and fourth ventricle ChP tissue were carefully microdissected and collected in serum-free RPMI at 4 °C. As ChP tissue is small and does not allow for single mouse scRNA-seq analyses, we pooled ChP tissues from eight mice to generate single pooled samples for naive 7-week-old (female), 22-week-old (male) and 82-week-old (male) mouse groups, for the lateral and fourth ventricle ChP tissue separately. After tissue collection, samples were pre-cut with small scissors and diluted and digested in 2:3 stock solution of collagenase I (10 U ml−1, Worthington), collagenase IV (400 U ml−1, Worthington) and DNase I (30 U ml−1, Worthington). Incubation was at 37 °C for 30 min with up and down pipetting every 10 min. Samples were strained through a 70-µm filter (Falcon) and washed with a total of 700 µl RPMI. Samples were centrifuged for 7 min at 300g at 4 °C, the supernatant was discarded and the pellet was resuspended in 250 µl FACS buffer (1× HBSS without Ca2±Mg2+, 2 mM EDTA and 2% BSA). Single live cells were sorted using the BD FACS ARIAII and loaded onto a Chromium GemCode Single Cell Instrument (10x Genomics) to generate single-cell gel beads in emulsion (GEMs). Libraries were prepared with the GemCode Single Cell 3′ Gel Bead and Library Kit v2 (10x Genomics) and the Chromium i7 Multiplex kit (10x Genomics) according to the manufacturer’s instructions. Afterwards, 10x barcoded cDNA was prepared for Illumina HiSeq4000 Next Gen Sequencing. Sequencing was performed at the VIB Nucleomics Core (VIB, Leuven). Demultiplexing of the raw data was carried out with the 10x Cell Ranger software (v.2.0.1). Obtained reads from demultiplexing were used as input for cellranger count to align reads to the mouse reference genome using STAR and collapse them to unique molecular identifier counts. The resulting expression matrices were used for follow-up analyses, which are described in the materials and methods section. After processing, we ended with 11,075 cells of pooled lateral vebtricle (LV) and fourth ventricle (4V) ChP samples of 7-week-old mice, 17,286 cells of pooled LV and 4V ChP samples of 22-week-old mice; and 16,294 cells of pooled LV and 4V ChP samples of 82-week-old mice. The tissue processing and sc/nRNA-seq analyses of embryonic and human ChP samples are described by Dani et al.5 and Yang et al.39, respectively.
sc/nRNA-seq merged datasets and analyses
We processed and combined various scRNA-seq and snRNA-seq datasets. The details and rationale for the processing pipelines are described below. Our custom pipelines can be viewed on GitHub at https://github.com/VandenbrouckeLab/Base_barrier_cells.
In-house individual ChP scRNA-seq objects
In total, there are six in-house ChP scRNA-seq samples used in this manuscript. This consists of a 4V and a LV ChP sample from 7-week-old, 22-week-old and 82-week-old mice. Each sample was first analyzed on its own according to the workflow listed below.
The data were pre-processed using the scater R package (v.1.14.6) according to the workflow proposed by McCarthy and colleagues52. Quality control metrics were calculated for all the single cells of the dataset using the function calculateQCMetrics. Outlier cells were identified based on three calculated metrics: library size, number of expressed genes and mitochondrial proportion. Cells were tagged as outliers if one of their three metrics was a minimum number of median absolute deviations (MADs) removed from the respective metric median for the dataset. For library size and number of expressed genes, this was 4 MADs lower or higher than the median. For mitochondrial proportion, only an upper limit was chosen and it was set at 5 MADs. After this first round of outlier removal, the runPCA function from scater was used for an extra round of outlier detection and removal.
After the initial pre-processing was complete, a pipeline was run according to the documentation from Seurat (v.3.1.4)53. First, the remaining good-quality cells were used to create a Seurat object. This was completed with the function CreateSeuratObject from Seurat with parameters min.cells = 3 and min.features = 200. The log normalization of the raw counts was performed with the NormalizeData function from Seurat with the scale.factor set to 10,000. Highly variable gene detection was completed with the FindVariableFeatures function from Seurat according to the vst selection method. The number of features was set to 2,000. Subsequently, the scRNA-seq data were scaled with the ScaleData function from Seurat. Then, principal-component analysis (PCA) was run with the RunPCA function from Seurat to identify the top 50 principal components (PCs). Finally, various number of PCs for dimensionality reduction and resolutions for unsupervised clustering were attempted. This was guided by running the ElbowPlot function from Seurat.
Doublets were removed based on DoubletFinder (v.2.0.2) prediction results and quality metrics54. If needed, some additional manual clustering was performed to split up certain clusters based on marker expression. Marker genes per identified subpopulation were found using the FindAllMarkers and FindMarkers functions of the Seurat package.
Seven-week-old ChP LV and 4V aggregate object
The aggregation of two in-house samples, 7-week-old mice ChP LV and 4V datasets, was carried out using ‘cellranger aggr’. This equalizes the average read depth per cell between the two samples before merging to avoid artifacts that may be introduced due to differences in sequencing depth. The post-normalization mean reads per cell of the aggregate object was 15,100 reads, as calculated by Cell Ranger. The subsequent aggregate dataset was pre-processed and analyzed according to the workflow explained above.
Twenty-five PCA dimensions and a resolution of 0.8 were used to create the final UMAP dimensionality reduction plot and clustering. The final object shown in this manuscript contains 11,075 cells and eight annotated clusters.
Marker analysis was performed on unsupervised clusters to get feature marker genes for each ChP cell type versus all other ChP cell types (Supplementary Table 1).
Functional annotation 7-week-old ChP LV and 4V aggregate object
GO enrichment analysis was performed using the clusterProfiler R package (v.3.14.3). Three ontologies (‘Biological Pathway’, ‘Cellular Compartment’ and ‘Molecular Function’) were included and a P value cutoff of 0.05 was utilized. All the genes from this scRNA-seq Seurat object were used as the background genes for the analysis. This analysis was conducted on the marker gene sets for ChP stromal and base fibroblasts. These gene sets were acquired by running the FindMarkers function separately for ChP stromal and base fibroblasts comparing each of them to all other non-fibroblast cell types. Only the positive logFC genes of the two resulting gene sets were taken into account for the subsequent GO enrichment analysis for both fibroblast subtypes. The results were then combined into one dot plot by running the merge_result function from clusterProfiler. The top 15 significantly enriched GO categories per ontology and per cell type are featured in the dot plot (owing to an overlap in the top GO categories, this amounts to fewer than 30 categories each). These top GO categories are ordered according to adjusted P values, prioritizing stromal fibroblasts. The adjusted P value is displayed as the color of the dot and the size of the dot is determined by the GeneRatio parameter, which is the ratio of the input DEG set annotated in the respective GO term.
Fibroblast origin complete object
To investigate the ontogeny of the type II fibroblasts we integrated some of our scRNA-seq datasets with public scRNA-seq datasets containing various ependymal, vascular and mesenchymal cell types from the brain.
|
Name |
Cell type |
Age |
Species |
Origin/paper |
|---|---|---|---|---|
|
GSM_2677817 |
Ependymal cells |
8 weeks |
Mouse |
Shah et al.12 |
|
GSM_2677818 |
Ependymal cells |
8 weeks |
Mouse |
Shah et al.12 |
|
GSM_2677819 |
Ependymal cells |
8 weeks |
Mouse |
Shah et al.12 |
|
Brain vascular cells |
10–19 weeks |
Mouse |
Vanlandewijck et al.7 |
|
|
ChP 4V_7w ChP LV_7w ChP_4V_22w ChP_LV_22w |
ChP vascular-associated cells |
7 weeks 7 weeks 22 weeks 22 weeks |
Mouse |
Current study |
|
Meningeal fibroblasts |
E14 |
Mouse |
Desisto et al.9 |
|
|
l6_r3_vascular_cells |
CNS vascular cells |
6–8 weeks |
Mouse |
Zeisel et al.11 |
|
l6_r4_ependymal_cells |
Ependymal cells |
6–8 weeks |
Mouse |
Zeisel et al.11 |
|
Meningeal fibroblasts |
Unknown 2–14 months Unknown |
Mouse |
Pietilä et al.10 |
For our four datasets, vascular and mesenchymal cells were selected based on the annotation in the individual processed Seurat objects of 4V and LV samples of 7- and 22-week-old mice. The selected cells were then combined into one dataset. The Cell Ranger aggregate method was not used here, in contrast to our 7-week-old object described above. In between both analyses it was determined, in other analyses in the BioIT core of our institute, that the Cell Ranger aggregate method did not provide any added benefit to downstream results and was no longer necessary. It was not used in the remainder of this manuscript.
For the public datasets, Shah et al.12 and Vanlandewijck et al.7, which did not provide a processed object, the datasets were pre-processed using the scater R package (v.1.14.6) as described above. Outlier cells were again identified based on the same three metrics, but now a cutoff of three MADs was used for each metric respectively. Additional outlier cell removal based on PCA was not needed. Subsequently, the beginning of the Seurat (v.3.1.4) pipeline was run for each dataset. First, the remaining good-quality cells were used to create a Seurat object. This was completed with the function CreateSeuratObject from Seurat with parameters min.cells = 3 and min.features = 200. The log normalization of the raw counts was performed with the NormalizeData function from Seurat with the scale.factor set to 10,000. Then, each Seurat object was saved.
The GSE150219 dataset from the Desisto et al. manuscript9 provided normalized counts and metadata post pre-processing, so no further pre-processing or normalization was needed. A Seurat object was created from the provided data and saved.
For the Zeisel et al. datasets11, we explored their Mouse Brain Atlas (http://mousebrain.org/). We downloaded two loom files containing cells relevant to our analysis, vascular cells annotated at taxonomy level 3 and ependymal cells annotated at taxonomy level 4 in their atlas. These datasets had been completely processed and were fully annotated, which meant that no further pre-processing was needed. The loom files just had to be converted to Seurat objects and saved.
For the three Pietilä et al. datasets10, we explored their processed data available at http://betsholtzlab.org/Publications/BrainFB/Data/BFBdata.html. We downloaded the raw count matrices and cell annotations from dataset 1, 2 and 4. These datasets had been pre-processed and were fully annotated. This meant no further pre-processing was needed. A Seurat object was created for each count matrix with no extra filtering. The log normalization of the raw counts was performed with the NormalizeData function from Seurat with the scale.factor set to 10,000. Their annotation was added as metadata and then each Seurat object was saved.
The data collection and pre-processing described above resulted in 11 individual Seurat objects. These objects were merged together using the merge function from Seurat.
This merged object was further analyzed according to the Seurat pipeline described above with one addition. After the RunPCA step, Harmony (v.1.0) batch correction55 was performed by running the RunHarmony function. This was completed with two batch variables, sample and dataset, supplied to the group.by.vars parameter. Each batch variable had a specified theta value provided to it, respectively 1 for the sample batch and 3 for the dataset batch. A higher theta value results in a stronger batch effect correction and as such a higher cluster diversity. This was carried out to integrate the various samples from the various datasets with each other in a measured way. It resulted in a good mixing of the respective cell types in the various datasets, so the datasets were deemed sufficiently integrated with Harmony.
Twenty Harmony-corrected PCA dimensions and a resolution of 0.8 were used to create the final UMAP plot and clustering.
The metadata of the individual objects was used to inform the annotation process. Additionally, the annotation was confirmed by performing automatic annotation with the R package ScType56. To achieve an optimal automatic annotation, a custom marker file was created by gathering marker lists from various brain single cell atlases and retaining the most frequent markers for each relevant cell type (CellMarker 2.0 database57, Azimuth Mouse Motor Cortex ref. 58,59, Mousebrain.org Adolescent Mouse Brain Atlas, via the UCSC Cell Browser60, and marker analysis from our data (Supplementary Table 1)). The final object contains 32,720 cells, 22 numbered clusters and 6 annotated clusters.
Fibroblast origin subset object
Following this initial analysis to confirm the fibroblast origin of our type II fibroblasts, we created a subset with only the fibroblast cluster (numbered clusters 1, 2, 5, 8, 14 and 20) to accurately compare the various fibroblasts across the datasets. Cells from the fibroblast cluster which clustered with the other cell types on the UMAP were also removed.
After the subset, the same workflow was performed as with the complete object above except for the batch correction step. Harmony was no longer performed due to it being insufficient to achieve optimal mixing of the datasets at the fibroblast subset level. Instead, Seurat v4 canonical correlation analysis (CCA) was performed; however, certain dataset batches did not have enough cells to be able to perform the CCA workflow. These datasets had to either be removed or had to be combined with other datasets into 1 batch (if this was applicable). The Shah et al. ependymal datasets12 and Zeisel et al. ependymal dataset11 had to be removed. The Vanlandewijck et al. vascular dataset7 was combined with the Pietilä et al. datasets10 1 and 2 into 1 batch because they are all 10x Genomics data, generated by the Betsholtz laboratory. The Desisto et al. dataset9, Zeisel et al. vascular dataset11, Verhaege et al. dataset and Pietilä et al. SmartSeq2 dataset10 all had enough cells and were kept as separate batches. This meant that the CCA workflow was performed on five batches in total. Fifteen PCA dimensions were used to find anchors and integrate the datasets. Ten integrated PCA dimensions and a resolution of 0.4 were used to create the final UMAP plot and clustering. It is noteworthy that this integrated assay was not used for the visualization of gene expression using dot plots and feature plots, for this the standard log-normalized RNA assay was used.
With the greater level of detail in this fibroblast subset some lingering contaminating endothelial and mural cells were identified and removed. The final object contains 11,076 cells and eight unsupervised numbered clusters. When the initial annotation from the metadata of the original individual objects was cleaned up and featured, there were 17 annotated clusters.
We validated our cell type annotation through automatic annotation with the previously created custom marker list detailed in the section before.
Marker analysis dot plot fibroblast origin subset object
Marker analysis via the Seurat function FindAllMarkers was performed using the revised (cleaned up) initial annotation, which stems from the original individual datasets. We wanted to find markers which were highly expressed in the various clusters, so the min.pct parameter was set at a very strict level of 0.6 to avoid lowly expressed markers. Next a filtering step was performed on this marker list to remove contaminating ChPE cell markers from our two fibroblast populations (utilizing the ChPE marker list acquired from the 7-week-old ChP LV and 4V aggregate object featured in Fig. 1b). This resulted in a strict filtered marker list for each cluster which was sorted according to the score column. Subsequently, top markers were manually selected from these lists to show the similarities and differences between the various fibroblast clusters and these genes were visualized in a dot plot.
Functional annotation fibroblast origin subset object
GO enrichment analysis was performed using the clusterProfiler R package (v.3.18.1). Two ontologies (‘Biological Pathway’ and ‘Cellular Compartment’) were included and a P value cutoff of 0.05 was utilized. All the genes from our scRNA-seq Seurat object were used as the background genes for the analysis. This was conducted on the marker gene sets for ChP stromal and base fibroblasts. These gene sets were acquired by running the FindMarkers function between ChP stromal and base fibroblasts in the final fibroblast origin subset object utilizing the revised original individual annotation. The resulting gene set was split up based on logFC, where positive logFC genes were linked to ChP stromal fibroblasts and negative logFC genes to ChP base fibroblasts. GO enrichment analysis was run on the two respective gene lists. The results were combined into one dot plot by running the merge_result function from clusterProfiler. The top 25 significantly enriched GO categories per ontology and per cell type are featured in the dot plot (owing to overlap in top GO categories for BP and the limited number of significant GO categories for CC, this amounts to fewer than 50 categories each). These top GO categories were ordered according to adjusted P value, prioritizing stromal fibroblasts. The adjusted P value is displayed as the color of the dot and the size of the dot is determined by the GeneRatio parameter, which is the ratio of the input DEG set annotated in the respective GO term.
ChP Fibroblast age object
To analyze how the transcriptome of fibroblasts varies with age, we integrated ChP scRNA-seq samples of differently aged mice: embryonal, 7 weeks old, 22 weeks old and 82 weeks old.
The 7-, 22- and 82-week-old mice ChP LV and 4V datasets were our own datasets. These six datasets were analyzed individually according to the pipeline described previously and the annotation was used to subset the fibroblast cells from the raw data of each dataset. Thereafter, all these fibroblast cells were combined into one Seurat object.
The ChP scRNA-seq embryonal data from the Lehtinen laboratory5 was acquired via the Single Cell Portal from the Broad Institute (https://singlecell.broadinstitute.org/single_cell/study/SCP1365/choroid-plexus-cell-atlas). The raw and normalized counts were provided together with some metadata and dimensionality reduction coordinates. This was used to create a Seurat object which was then subsequently subsetted to only include the mesenchymal cells. Last, the third ventricle cells were removed from the object because our datasets do not include the third ventricle.
Following this initial prep, these two Seurat objects were integrated via Seurat v4 CCA according to the scRNA-seq integration procedure from Seurat with the functions SelectIntegrationFeatures, FindIntegrationAnchors and IntegrateData (https://satijalab.org/seurat/articles/integration_introduction.html) using default parameters. This resulted in an integrated data assay which was used as the default assay for the rest of the Seurat workflow as described previously. One exception though was the visualization of the expression of typical cell type markers using violin plots and feature plots where the standard log-normalized RNA assay was used.
Thirty integrated PCA dimensions and a resolution of 0.8 were used to create the UMAP plot and clustering. Some additional manual clustering was performed to split up certain clusters based on marker expression. Among our initially selected fibroblast cells there were, however, some contaminating cells, which clustered separately and were removed. Additionally, the mesenchymal cells from the Dani et al.5 dataset contained some nonfibroblast cells and proliferating fibroblasts, so these clusters were also removed. The final object contains 3,116 cells and four annotated clusters.
Stacked bar chart ChP fibroblast age object
The contribution from the various ages to the two fibroblast populations of interest was analyzed and visualized with a percentage stacked bar chart. The difference in total cells between the four ages was taken into account by comparing relative amounts for each cell population instead of absolute numbers.
Differential expression analysis ChP fibroblast age object
Differential expression analysis (DEA) between the mouse age categories was performed via the Seurat function FindMarkers. The test.use parameter was set to MAST (model-based analysis of single-cell transcriptomic)61. This method determines DEGs via the hurdle model between two groups of cells and is customized for scRNA-seq data. This was conducted to assess how much the transcriptome of ChP stromal and base fibroblasts changes across the ages of the mice.
ChP fibroblast species object
To check the cross-species nature of our mouse ChP base fibroblasts, we looked for a human dataset of the ChP containing a sizable mesenchymal cell proportion. The snRNA-seq dataset from the Yang et al.39 study fitted the bill. The human Choroid Plexus Seurat object was acquired through the Stanford Shiny app (https://twc-stanford.shinyapps.io/scrna_brain_covid19/) as an rds file. This was the analyzed object with all the metadata, so no pre-processing was needed. Applicable human fibroblast markers were visualized to confirm the identity of the mesenchymal cell cluster to then subset the object to include only the mesenchymal cells to integrate with our mouse datasets.
For this analysis we used the same six datasets from our laboratory, which we used in the previous fibroblast age object analysis, but the merged mouse object was now first analyzed completely on its own so that contaminating cells could be removed before integration.
Integrating the merged object from our six mouse datasets with the mesenchymal subset of the human dataset proved to be a challenge. As we were integrating scRNA-seq with snRNA-seq across species, it was necessary to apply a stronger batch effect correction method than before. After studying a recent benchmark of integration methods62, we chose to implement the batch-balanced k-nearest neighbors (BBKNN) algorithm63 to achieve an optimal result.
Both Seurat objects were updated through the UpdateSeuratObject function to be able to analyze it further in a newer R environment (v.4.0.5) and Python environment (v.3.9.9) for BBKNN (v.1.5.1). Before integration via BBKNN, the gene symbols of the human mesenchymal subset first had to be converted to their mouse orthologs. This was accomplished by utilizing the Ensembl database through the biomaRt package (v.2.46.3). Human genes without mouse orthologs were removed from the count matrix. A new human Seurat object was created from this converted raw count matrix. Metadata from the original object was transferred to this new human Seurat object. When merging the human mesenchymal dataset with our merged mouse dataset, the count matrices were subsetted to the common genes between the two datasets.
Subsequently, a part of the previously described Seurat workflow was run in R on the merged cross-species object. Log normalization of the raw counts was performed with the NormalizeData function from Seurat with the scale.factor set to 10,000. Highly variable gene detection was completed with the FindVariableFeatures function from Seurat according to the vst selection method. The number of features was set to 2,000. Subsequently, the scRNA-seq data were scaled with the ScaleData function from Seurat. Then, PCA was run with the RunPCA function from Seurat to identify the top 30 PCs.
Thereafter, the cross-species Seurat object was converted to an AnnData python object to run BBKNN in Python. Before running BBKNN, ridge regression was used with the species metadata as the batch effect and the initial Leiden clustering as the biological grouping, which should improve the integration. BBKNN was then run on the first 30 PCA dimensions. The 30 new integrated PCA dimensions were then used to determine a corrected UMAP dimensionality reduction with a clustering resolution of 0.5.
After the Python analysis was completed, the AnnData object was converted back into a Seurat object for further downstream analysis in R. The final object contains 5,406 cells and eight annotated clusters.
Stacked bar chart ChP fibroblast species object
The contribution from the two species to the eight annotated fibroblast clusters was analyzed and visualized with a percentage stacked bar chart. The difference in total cells between the two datasets was taken into account by comparing relative amounts for each cell population instead of absolute numbers.
Conserved gene expression analysis dot plot ChP fibroblast species object
To determine the genes which are conserved between human and mouse for the ChP base fibroblast population, the FindConservedMarkers function from Seurat was run on the ChP base fibroblast cluster (versus all other cells) with species as the grouping variable and otherwise default parameters. This returned a ranked list of putative conserved genes with the associated statistics for each species and also a couple of summary columns. This list of conserved genes was ordered according to ascending max P value (largest P value between both species) and the top five genes with a mouse log2FC > 1.5 were selected to feature in the dot plot of conserved genes.
Statistical analysis markers
DEA to determine the cluster markers was performed using the Wilcoxon rank-sum test through the Seurat functions FindAllMarkers and FindMarkers. RNA markers for the annotated clusters of ChP LV and 4V aggregate were determined with the cutoffs min.pct = 0.10, min.diff.pct=0.25, logfc.threshold = 0.30, return.thresh = 0.01 and only positive markers were evaluated. For most of the other objects, markers were determined with default cutoffs of min.pct = 0.10, min.diff.pct = -Inf, logfc.threshold = 0.25, return.thresh = 0.01, only.pos = FALSE; except for the fibroblast origin subset object, where the min.pct was set at a strict 0.6 to find highly expressed markers and logfc.threshold was set at 0.30 in the marker analysis on which GO enrichment analysis was run. An extra ‘score’ column was always calculated as a way to rank the importance of the genes as markers. It was calculated with the function ‘pct.1/(pct.2 + 0.01) × avg_logFC’. The markers were ordered according to this score.
DEA to determine the DEGs between the mouse age categories in the ChP fibroblast age object was performed using MAST through the Seurat function FindMarkers. This was run with a logFC.threshold = 0 to make volcano plots and min.pct was set to 0.10 to retain only genes that are meaningfully expressed throughout the cell population.P value adjustment for DEA was always accomplished with Bonferroni correction.
Electron microscopy
Mice for EM analyses were sedated as described above and brains were isolated and fixed in 2% PFA with 2% glutaraldehyde for 1 h at room temperature, followed by overnight incubation at 4 °C. After thorough washing with PBS, brains were embedded in 5% agarose and 100–200-µm thick sections were cut with a Leica Vibratome VT1200S. Sections with a clear ChP base region were selected and further cut into small squares. Tissues were processed for SBF-SEM analyses with the OTO protocol as described previously64 with the sole exception that we did not use Spurr’s resin but used Embed 812. Samples were mounted onto aluminum pins and trimmed and SBF-SEM imaged with the Zeiss Merlin with 3View2 (Gatan). When desired, regions with clear ChP base region were also transferred to and imaged with a Zeiss Crossbeam 450 for FIB-SEM imaging65. For tracing we used Microscopy Image Browser software and 3D rendering was conducted in Imaris. For TEM analyses, ultrathin sections of a gold interference color were cut using an ultra-microtome (Leica EM UC7), followed by post-staining with uranyl acetate and lead citrate in a Leica EM AC20 and collected on formvar-coated copper slot grids. They were viewed with a JEM 1400plus transmission electron microscope (JEOL) operating at 80 kV.
Correlative light electron microscopy
A CLEM workflow was performed as described elsewhere26,66,67. In brief, mice were i.v. injected with 1 mg 200 µl−1 20 g−1 BW ConA-FITC and immediately sedated with a mix of ketamine (20 mg ml−1) and xylazine (4 mg ml−1). After 5 min, mice were decapitated and brains were sampled and fixed in 4% PFA with 0.25% glutaraldehyde for 4 h at 4 °C. Brains were washed with PBS and embedded in 5% agarose and cut into 50 µm-thick sections using a Leica Vibratome VT1200S. We performed free-floating anti-CLDN11 immunostaining on these sections. After mounting, samples were first scanned for regions of interest (ROIs) and branded using Near Infra-Red Branding using a Zeiss LSM780 with MaiTai laser68. ROIs were then imaged through LSM880 FastAiry super resolution imaging. These same samples were then processed for volume EM as described above. Overlaying both confocal light microscopy and volume EM datasets was carried out with the Bigwarp plugin of ICY69 and using branching points of the blood vessels as points for landmark-based registration.
Statistical analyses
No statistical methods were used to predetermine sample sizes but our sample sizes were similar to those reported in previous publications. For most experiments we used a mix of both male and female mice, unless stated otherwise. Statistical analysis was conducted using Prism v.9.3.1. Dotplots show average value per mouse ± s.d. Normal distribution of data was checked using the Kolmogorov–Smirnov test and Shapiro–Wilk tests. For CLDN11-AI14 colocalization analysis, data were normally distributed according to Shapiro–Wilk test and a Welch’s t-test was performed. For CLDN11–dextran colocalization analysis, data were not normally distributed and we therefore performed the strictest Mann–Whitney U-test. For ZO1/CDH1 intensity analyses, data were not normally distributed and therefore the strictest Mann–Whitney U-test was performed. For qPCR analysis, data were normally distributed after log-transformation and we therefore performed unpaired t-tests. Differences in CCR2+ cells per volume of base versus central regions in naive and systemically inflamed mice was tested using a two-way ANOVA (Sidak). Statistical tests performed on scRNA-seq data (for example through gene set enrichment analysis) were corrected for multiple testing. Details for scRNA-seq-related tests and statistics are described in Methods.
Inclusion and ethics
Animal studies (EC2020-005, EC2021-048, EC2022-004 and EC2023-039) were approved by the ethical committee of Ghent University Center for Inflammation Research, Belgium. Embryonic C57BL/6J mouse work was performed in accordance with the Institutional Animal Care and Use Committee and relevant guidelines of Boston Children’s Hospital and Masaryk University. This study did not involve human participants or patient recruitment, and no demographic variables were used. Human samples were obtained under an IRB-approved protocol at Boston Children’s Hospital. A neuropathologist (H.G.W.L.) reviewed the human tissue specimens and recorded nonidentifying information. No statistical methods were used to predetermine sample sizes but our sample sizes are similar to those reported in previous publications.
Additional information
Additional information is available at https://doi.org/10.5281/zenodo.14703050 (ref. 35). 1. RNA markers numbered clusters CNS merged dataset: RNA markers for the numbered clusters of the CNS vascular-associated and glial cell merged scRNA-seq dataset. Marker genes were determined by DEA for each cluster versus all other clusters. This was performed by running the Seurat function FindAllMarkers (Methods). 2. Automatic annotation ScType custom markerfile: Custom marker file, created by exploring various brain single cell atlases, which was used to automatically annotate the cell populations in the CNS vascular-associated and glial cell merged scRNA-seq dataset. 3. RNA markers numbered clusters FB subset of CNS merged dataset. RNA markers for the numbered clusters of the CNS fibroblast subset dataset: Marker genes were determined by DEA for each cluster versus all other clusters. This was performed by running the Seurat function FindAllMarkers (Methods). 4. RNA markers annotated clusters ChP fibroblast age dataset: RNA markers for the manually annotated clusters of the dataset consisting of ChP fibroblasts of E16, 7-, 22- and 82-week-old mice. Marker genes were determined by DEA for each cluster versus all other clusters. This was performed by running the Seurat function FindAllMarkers (Methods).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.