
This study successfully established a murine model of obesity-associated asthma by combining high-fat diet (HFD)-induced obesity with ovalbumin (OVA)-induced asthma. We investigated the effects of the AMPK activator metformin and explored the role and mechanism of the AMPK/NLRP3 signaling pathway. Our findings demonstrate that metformin intervention activates the AMPK signaling pathway, inhibits the activation of the NLRP3 inflammasome and its downstream effector Caspase-1, consequently reduces levels of pro-inflammatory cytokines IL-1β, IL-18, and leptin, and ameliorates airway inflammation in obesity-associated asthmatic mice.
Obesity represents a state of low-grade chronic inflammation associated with alterations in gut microbiota, cellular metabolism, lipid handling, immune function, insulin resistance, and adipokine production by adipose tissue21. Two primary hypotheses link obesity and asthma: one involves mechanical effects such as diaphragmatic displacement due to fat deposition and reduced chest wall compliance22, while the other emphasizes the role of adipose tissue-derived immunomodulatory and inflammatory adipokines, such as leptin and adiponectin23. Leptin, a key pro-inflammatory adipokine, exacerbates airway pathology through multiple immunoregulatory mechanisms. Studies indicate leptin promotes CD4 + T cell differentiation towards Th1/Th17 phenotypes via the JAK2-STAT3 pathway, upregulates pro-inflammatory cytokine secretion, and simultaneously suppresses Th2 cytokines and regulatory T cell (Treg) function. This results in increased Th1 cell infiltration and decreased Treg proportion in the lung tissue of obese asthmatic mice, aggravating airway inflammation24. Wang et al. reported that leptin significantly upregulates JAK2-STAT3 phosphorylation in obese asthmatic mice, inducing M1 macrophage polarization and promoting the expression of M1 markers (iNOS, IL-6, TNF-α), while suppressing M2 markers (Arg-1). This polarization shift showed a significant positive correlation with airway wall collagen deposition scores, directly linking it to airway remodeling and suggesting its involvement in structural airway alterations20. Marek et al. demonstrated that leptin facilitates NLRP3 inflammasome assembly by upregulating TXNIP, enhancing Caspase-1 activation and IL-1β maturation/release. This process was effectively blocked by TXNIP-specific inhibitors or siRNA, resulting in significantly reduced NLRP3 assembly and markedly decreased IL-1βsecretion25. Furthermore, leptin induces reactive oxygen species (ROS) generation by activating oxidative stress pathways, establishing a “leptin–oxidative stress–inflammasome” positive feedback loop26. Thus, leptin acts as a core driver of metabolic-immune imbalance in obesity-associated asthma by regulating multiple pathological mechanisms, including T-cell polarization, macrophage activation, inflammasome triggering, and oxidative stress. Targeting leptin represents a promising therapeutic strategy for developing novel interventions for this asthma phenotype.
AMPK, a pivotal hub integrating energy metabolism and inflammation regulation, plays a critical role in the pathogenesis of obesity-associated asthma. Suppression of its activity exacerbates disease progression. Xia et al. revealed that reduced AMPK phosphorylation in obesity stems from an imbalance in the AMP/ATP ratio caused by HFD-induced mitochondrial dysfunction, directly inhibiting AMPK activation27. Research has confirmed that in the obese state, elevated leptin levels coincide with decreased receptor sensitivity28. This leads to sustained JAK2 phosphorylation and consequent STAT3 activation. Phosphorylated STAT3 directly binds to the promoter region of the LKB1 gene, suppressing its transcriptional expression. As LKB1 is the upstream kinase of AMPK, its impaired activity directly blocks phosphorylation of the AMPKαsubunit at Thr172, resulting in reduced AMPK activity29. Conversely, AMPK activation significantly improves obesity-associated asthma. Zhu et al., in studies using an obese asthmatic mouse model, demonstrated that AMPK activation attenuates airway inflammation by inhibiting the NF-κB pathway. This inhibition reduces the expression of pro-inflammatory factors (such as TNF-α and iNOS) and the anti-apoptotic protein Bcl-2, thereby suppressing inflammatory cell recruitment and apoptosis resistance. Furthermore, AMPK activation alleviates the oxidative microenvironment in obese asthmatic mice. It achieves this by inhibiting iNOS-mediated nitric oxide (NO) production, reducing levels of the oxidative stressmarker 8-hydroxy-2’-deoxyguanosine (8-OHdG), and enhancing total antioxidant capacity, thus mitigating oxidative damage to the airway and vascular endothelium30. Lei et al. demonstrated that AMPK activation, via the JAK2/STAT3 pathway, blocks transcription of M1 macrophage polarization-related genes while promoting M2 macrophage marker expression, reducing the secretion of pro-inflammatory cytokines (TNF-α, IL-6, IL-1β). Ultimately, these effects reduce airway inflammatory cell infiltration, goblet cell hyperplasia, and collagen fiber deposition, leading to improved airway hyperresponsiveness (AHR) and attenuated airway remodeling in obese asthmatic mice31. Furthermore, AMPK activation promotes Nrf2 nuclear translocation and upregulates HO-1 expression, concurrently inhibits the NF-κB pathway by reducing p-IκBα and p65 nuclear translocation, decreases pro-inflammatory cytokines (TNF-α, IL-1β), enhances antioxidant enzyme activity (SOD, GSH, CAT), and lowers MDA and ROS levels, thereby ameliorating airway inflammation and remodeling in asthmatic mice32. Collectively, these mechanisms establish AMPK as a central nexus connecting energy metabolism and inflammation, highlighting its importance as a therapeutic target for obesity-associated asthma.
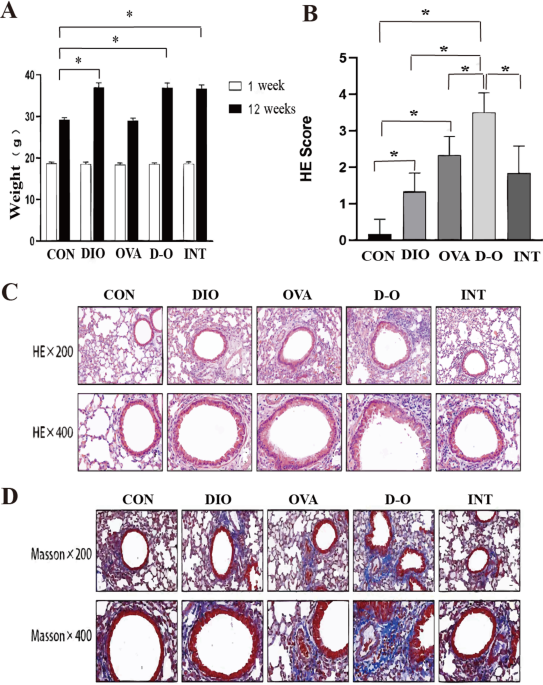
Concurrent inhibition of AMPK signaling and activation of the NLRP3 inflammasome drive the inflammatory cascade in obesity-associated asthma. Excessive NLRP3 inflammasome activation leads to caspase-1 cleavage/activation and the maturation/release of pro-inflammatory cytokines IL-1β and IL-189. Substantial evidence indicates that NLRP3 inflammasome activation can be inhibited by targeting key upstream or downstream components. Zhang et al. demonstrated that GLCCI1 inhibits NLRP3 inflammasome activation by suppressing the PI3K pathway. This inhibition reduces the protein expression of NLRP3, ASC, and Caspase-1, along with the secretion of IL-1β and IL-18, thereby ameliorating airway inflammatory cell infiltration, goblet cell hyperplasia, and collagen deposition in asthmatic mice. Consequently, airway hyperresponsiveness (AHR) and airway resistance are improved33. Liu et al. discovered MUC1 binds to TLR4, impeding its interaction with MyD88, thereby inhibiting NLRP3 inflammasome activation, downregulating NLRP3, Caspase-1, IL-1β protein expression, and alleviating airway inflammation in asthmatic mice34. In our experiment, IL-1β and IL-18 levels were significantly higher in the obese asthma group compared to controls and positively correlated with airway inflammation scores. These results further confirm that the AMPK-NLRP3 pathway serves as a core hub linking dysregulation of energy metabolism to activation of the innate immune response.
Metformin, a classical AMPK activator, exerts therapeutic effects in obesity-associated asthma through multiple mechanisms. Relevant studies suggest metformin inhibits mitochondrial respiratory chain complex I, disrupting electron transport, leading to AMP and ADP accumulation and ATP depletion, thereby indirectly activating AMPK35. In addition, metformin activates AMPK, resulting in mTOR phosphorylation and inactivation. This suppresses S-phase kinase-associated protein 2 (Skp2) expression, preventing Skp2-mediated degradation of cyclin-dependent kinase inhibitor 1B (p27). Consequently, p27 inhibits airway smooth muscle cell proliferation, thereby suppressing airway remodeling and preventing irreversible airflow obstruction36. Our study confirmed that metformin intervention significantly increased p-AMPK protein expression while decreasing NLRP3 and Caspase-1 protein levels, and downregulated serum leptin, IL-1β, and IL-18 levels in obese asthmatic mice. Histopathology revealed metformin treatment significantly reduced bronchial wall thickness, collagen deposition area, and total leukocyte count along with neutrophil/eosinophil proportions in bronchoalveolar lavage fluid (BALF). This demonstrates that metformin attenuates airway inflammation and remodeling, underscoring the crucial role of AMPK activation by metformin in mitigating obesity-associated asthma.
Although this study establishes the central role of the AMPK-NLRP3 pathway in obesity-associated asthma, limitations exist. Given species differences between mice and humans, further investigation using patient samples and data is warranted. This will elucidate whether metformin confers similar protective effects in humans and inform better therapeutic strategies for obese asthmatic patients. In this study, OVA was used to induce asthma, a model widely employed in allergic asthma research due to its high reproducibility37,38. However, OVA is not the only agent used for asthma induction; other stimuli such as house dust mite (HDM) extract and cockroach allergens can also elicit asthmatic responses39,40. The HDM-induced asthma model better recapitulates the chronic features and heterogeneity of human asthma, including corticosteroid resistance. Obesity was induced in this study using a high-fat diet (HFD), which represents a standard model for studying obesity-related diseases41. It should be noted, however, that Western diets are typically characterized by high sugar content, and excessive sugar intake can directly contribute to adipose tissue accumulation and metabolic inflammation42. Compared to high-fat diets, high-sugar diets promote obesity through distinct mechanisms and may elicit different inflammatory responses in the airways. Therefore, future studies could employ high-sugar diet-induced obesity models to examine whether the modulatory effects of metformin on the AMPK/NLRP3 signaling pathway and airway inflammation remain consistent. Furthermore, this study demonstrates that metformin inhibits the upstream NLRP3/Caspase-1 pathway in the lungs and concurrently reduces serum IL-1β and IL-18 levels, but lacks direct measurement of these cytokines in bronchoalveolar lavage fluid or lung homogenates. Future research directly quantifying active IL-1β and IL-18 levels in the pulmonary microenvironment will provide more definitive evidence for the local anti-inflammatory effects of metformin. Finally, although the sample size in this study was sufficient to detect significant effects, future investigations employing larger cohorts (e.g., n = 8 to 10 per group) would provide even greater statistical power and reinforce the generalizability of these findings.
Airway inflammation in obesity-associated asthma results from the interplay of metabolic dysregulation, aberrant signaling pathways, and cytokine imbalance. Within this context, suppressed AMPK signaling and concomitant NLRP3 inflammasome activation represent a central pathogenic hub. Metformin, by activating AMPK and suppressing the NLRP3/Caspase-1 pathway and downstream pro-inflammatory cytokine release, significantly ameliorates airway inflammation and pathological remodeling in obesity-associated asthma. This study not only delineates the pathogenic mechanisms underlying obesity-associated asthma in greater detail but also provides crucial experimental evidence supporting the development of AMPK-targeted therapeutics for this condition.