
The emergence of novel viral infectious diseases, such as the coronavirus disease 2019 (COVID-19) pandemic, has inflicted profound and devastating impacts on global health and socio-economic stability1. This war against the pandemic has not only exposed the limitations of human understanding of viruses but also served as a stark warning: in the enduring evolutionary contest between viruses and their hosts, viral evolution far outpaces our imagination. Confronted with this challenge, a deep dissection of host factors, particularly the pivotal role of long non-coding RNAs (lncRNAs, >200 nucleotides in length with no protein-coding potential) in virus–host interactions, is paramount for scientifically countering future threats from emerging viruses.
Early research on virus–host interactions focused on protein-mediated mechanisms. The discovery of lncRNA NEAT1’ role in regulating human immunodeficiency virus type 1 (HIV-1) mRNA transport unveiled a new lncRNA-mediated regulatory axis2. Subsequent studies have confirmed that host lncRNAs can exert potent antiviral effects by modulating gene expression, immune responses, or directly targeting viral genomes. However, the functionality of lncRNAs is akin to a double-edged sword; they can be hijacked by viruses to suppress host defense mechanisms (e.g., interferon response) and to facilitate viral replication and dissemination. Critically, the expression and function of lncRNAs significantly influence clinical progression and outcomes of viral diseases, highlighting their substantial potential as diagnostic biomarkers and therapeutic targets.
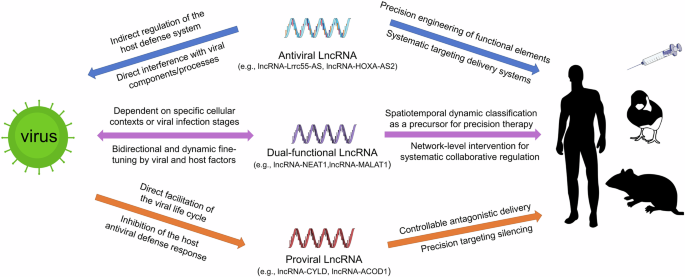
Given the intricate dual nature of lncRNAs during infection, we propose a framework (Fig. 1) categorizing them as: antiviral lncRNAs that participate in host antiviral defense, proviral lncRNAs that are exploited by viruses to promote infection, and bifunctional lncRNAs that act as environmentally sensitive switches. It should be emphasized that this tripartite classification represents a context-dependent summary of current experimental evidence, rather than an absolute, invariant biological property of individual lncRNAs. LncRNA function is highly plastic and frequently shaped by viral strain, host cell type, infection stage, and cellular signaling milieu. For many lncRNAs currently annotated as exclusively antiviral or proviral, their regulatory behavior may shift or reverse in untested experimental contexts, and future studies will likely uncover bifunctional activities that are not yet reported in the literature. This classification, therefore, serves as a heuristic framework for organizing existing findings while acknowledging the inherent limitations of incomplete experimental coverage. Therapeutic interventions targeting them must be designed with high specificity and precision based on this classification.

Schematic depicts host lncRNAs as antiviral defenders (e.g., lncRNA-Lrrc55-AS4, lncRNA-HOXA-AS210), proviral accomplices (e.g., lncRNA-CYLD18, lncRNA-ACOD127), or dual-functional arbiters (e.g., lncRNA-NEAT136,50, lncRNA-MALAT138,39) dynamically regulated by viral/host factors. The schematic also outlines integrated therapeutic strategies that enable function-specific targeting of lncRNAs via RNA engineering, spatiotemporal delivery, or network-based modulation to disrupt viral replication cycles.
This review systematically summarizes the diverse molecular mechanisms by which lncRNAs mediate virus–host interactions, evaluates their therapeutic potential and challenges, and provides a foundation for developing next-generation, highly precise and efficient antiviral therapies targeting host lncRNAs.
LncRNA constructs a multidimensional regulatory network in antiviral defense
The dual core functions of lncRNAs in immune defense
As strategic regulatory hubs within the host antiviral system, lncRNAs establish a dynamic defense framework by integrating innate and adaptive immune programming. In innate immunity, lncRNAs function across the entire spectrum—from pathogen recognition to effector response termination. For instance, lncRNA lnczc3h7a, which is significantly upregulated in response to viral infection, precisely regulates downstream signaling of pattern recognition receptors (PRRs): it serves as a molecular scaffold to bind tripartite motif containing 25 (TRIM25) and activated retinoic acid‑inducible gene I (RIG-I), and facilitates TRIM25-mediated K63-linked ubiquitination of RIG-I to activate downstream antiviral innate immunity3. As signaling cascades progress, lncRNAs exert critical control by directly targeting cascade components. For example, lncRNA lncLrrc55-AS is virus-inducible via the interferon-Janus kinase-signal transducer and activator of transcription (IFN-JAK-STAT) pathway. This lncRNA promotes the interaction between phosphatase methylesterase 1 (PME-1) and Phosphatase 2 A (PP2A), mediating demethylation of the PP2A catalytic subunit at Leu309 and consequent loss of PP2A phosphatase activity, which alleviates PP2A-mediated suppression of interferon regulatory factor 3 (IRF3) phosphorylation, thereby potentiating antiviral signaling4. At the effector stage, lncRNAs directly modulate key effector molecules to fine-tune downstream gene expression. For instance, porcine reproductive and respiratory syndrome virus (PRRSV) infection induces the upregulation of lncRNA LNC_000397, which negatively regulates PRRSV replication by promoting the expression of antiviral interferon-stimulated genes (ISGs), including MX dynamin-like GTPase 1 (MX1), ISG15, and radical S-adenosyl methionine domain containing 2 (RSAD2)5. Notably, lncRNAs also help maintain homeostasis; IFN-induced lncRNA lnc-Lsm3b competitively binds RIG-I to suppress excessive IFN production and prevent immunopathology6.This homeostatic function is firmly validated by in vivo data from lnc-Lsm3b-deficient mice, which display exaggerated type I IFN responses upon viral infection, supporting its physiological role in restricting overactive innate immunity. This hierarchical regulation enables precise spatiotemporal control of innate immunity (Fig. 2).

The schematic outlines the core framework of antiviral innate immune signaling: upon recognition of viral pathogen-associated molecular patterns by pattern recognition receptors (including TLR family, RIG-I/MDA5, and cGAS), the downstream signaling cascades are initiated to activate key transcription factors (NF-κB, IRF1/3/7/8, and STATs), which drive the production of IFN. Secreted IFNs bind to the interferon receptor (IFNAR) on the cell membrane to trigger the JAK-STAT signaling axis, further inducing the transcription of ISGs that establish the cellular antiviral state56,57,58,59. This figure depicts the positive regulatory roles of lncRNAs in the above core antiviral pathways (TLR, RIG-I/MAVS, and cGAS-STING pathways, as well as the IFN-JAK-STAT-ISG feedback loop). Representative lncRNAs (e.g., lncRNA-Lrrc55-AS4, lncRNA-CHROMR60) boost the signaling cascades at multiple regulatory nodes, amplify IFN and ISG responses, and ultimately restrict viral replication. Solid black lines with arrowheads indicate activation; solid black lines with T-bar ends indicate inhibition; red asterisks mark representative lncRNAs cited in this legend; red text indicates lncRNAs; colored boxes indicate core signaling proteins and pathway nodes. All abbreviations are defined in the Glossary.
This sophisticated regulation extends to adaptive immunity where lncRNAs orchestrate long-term defense. Notably, hepatitis B virus (HBV) infection markedly upregulates lncRNA lnc-DC expression in dendritic cells (DCs). Functionally, the upregulated lnc-DC sustains STAT3 phosphorylation downstream of toll-like receptor 9 (TLR9), modulates the cytokine secretion profiles, reducing tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), IFN-γ and IL-1β, and enhances antigen presentation by DCs, collaboratively promoting the formation of an immune microenvironment conducive to T cell activation7,8. Regarding T cell fate determination, lncRNA Snhg1 exhibits a highly temporally dynamic expression profile during virus infection-driven CD8+T cell differentiation, which follows a canonical naïvehi-effectorlo-memoryhi pattern tightly coupled to the memory fate commitment of CD8+T cells. Mechanistically, Snhg1 promotes the membrane localization of IL-7Rα by binding to the vesicular trafficking protein Vacuolar protein sorting 13 homolog D (Vps13D), thereby regulating cellular survival through the STAT5-B-cell lymphoma 2 (BCL-2) axis, and initiating the memory differentiation program via the STAT3-T cell factor 1 (TCF1)-B lymphocyte-induced maturation protein 1 (Blimp1) axis9. Through immunological memory programming, lncRNAs create defense continuity spanning viral clearance to immune surveillance.
Direct intervention mechanisms of lncRNAs on viral genomes
During the long-term co-evolution of viruses and hosts, host cells have evolved the ability to utilize lncRNAs to implement three core defensive strategies (Fig. 3). The most direct mechanism involves lncRNAs binding to viral genomes, including the genomic RNA of RNA viruses, genomic DNA of DNA viruses, and viral replication intermediates that serve as the genetic template for viral propagation. For example, lncRNA HOXA-AS2, which is significantly upregulated upon HBV infection, forms triplex structures with HBV covalently closed circular DNA (cccDNA) to recruit the metastasis-associated protein 1 (MTA1)-histone deacetylase 1/2 (HDAC1/2) deacetylase complex and mediates the epigenetic silencing of viral transcription10. Similarly, lncRNA ALPHA is significantly induced by chikungunya virus (CHIKV) infection, and directly binds to the genomic RNA of CHIKV to interfere with its replication process11. Critically, this direct targeting offers high specificity and inherent safety, new advantages for novel antiviral strategies.

Schematic depicts the viral replication cycle (attachment to release) and critical host pathways (e.g., ER stress, apoptosis). Antiviral lncRNAs (e.g., lncRNA-HOXA-AS210, lncRNA-ALPHA11) suppress infection at specific stages (e.g., entry blockade, replication suppression, protein synthesis disruption) and modulate stress/death pathways, thereby curbing viral replication and spread. Purple structures indicate viral particles. All abbreviations defined in the Glossary.
A second strategy entails precise modulation of host proteins. HBV-encoded DNA polymerase enhances the transcriptional activity of host factor cAMP-responsive element-binding protein 1 (CREB1) to drive the expression of lncRNA HOTTIP, which in turn upregulates Homeobox A13 (HOXA13) to directly bind the HBV genome and suppress viral transcription and replication12. Another key example is EDAL, a host lncRNA significantly induced by neurotropic virus infection, which specifically binds to the core catalytic subunit enhancer of zeste homolog 2 (EZH2) of the polycomb repressive complex 2 (PRC2), and shields its T309 site to block its O-GlcNAcylation13. This subsequently promotes the degradation of EZH2 via the lysosomal pathway, reduces histone H3 lysine 27 trimethylation (H3K27me3) modification levels, and thereby activates the transcription of the antiviral peptide purkinje cell protein 4-like 1 (PCP4L1). The intrinsic antiviral activity of PCP4L1 has been experimentally validated in this study, and its upregulation effectively inhibits viral replication. These mechanisms demonstrate that lncRNAs primarily exert their effects indirectly on viral genomes or activation of downstream antiviral effectors by finely regulating the stability, activity, or post-translational modifications of host proteins.
Thirdly, lncRNAs function as competitive endogenous RNAs (ceRNAs). A typical example of this regulatory mode during viral infection is lncRNA SNHG9, which is significantly upregulated upon enterovirus D68 (EV-D68) infection. It acts as a molecular sponge for miR-150-5p to relieve the microRNA (miRNA)-mediated suppression of the proviral transcription factor c-Fos, thereby facilitating viral replication14. This dynamic ceRNA regulatory mechanism enables lncRNAs to flexibly modulate key nodes in the host antiviral signaling network.
Notably, some lncRNAs integrate multiple antiviral mechanisms, exhibiting context-dependent plasticity. GAS5 is a prominent example: it is significantly upregulated upon hepatitis C virus (HCV) infection, and directly binds the HCV NS3 protein as a decoy to exert antiviral effects15; in sharp contrast, GAS5 expression is significantly suppressed by HIV infection, a change that cripples its two core intrinsic antiviral regulatory modes16,17. GAS5 normally functions as a ceRNA to competitively bind miR-873, attenuating the miRNA’s regulatory effect on target genes for viral suppression16, and modulates miR-21-mediated signaling to regulate T cell receptor (TCR)-driven CD4+T cell activation and apoptosis17, thereby influencing antiviral immunity. This single-agent, multi-target capacity reveals avenues for multi-target therapeutic design.
Despite these diverse antiviral functions, clinical translation faces bottlenecks including low cross-species conservation and insufficient in vivo delivery efficiency. More critically, viruses can hijack host lncRNAs for immune evasion (such as HIV-induced downregulation of the antiviral lncRNA GAS5), underscoring the complexity of host-virus interactions. Deep dissection of lncRNA defense networks and hijacking principles is crucial for developing novel antiviral targets and understanding evolutionary arms races.
LncRNAs as viral collaborators to subvert the host defense network
LncRNAs assist viruses in weaving a covert network for immune evasion
Viruses have evolved sophisticated mechanisms to evade immune responses by inducing the functional reprogramming of specific lncRNAs and suppressing antiviral lncRNAs. This strategy targets three core components of the host innate defense system: PRR surveillance, signal transduction and transcriptional activation, and the activity of antiviral effector proteins (Fig. 4).

Building on the core antiviral signaling cascade detailed in Fig. 2 (involving PRRs, downstream transcription factors, IFNs, IFNAR, the JAK-STAT axis, and ISGs), this schematic illustrates how viruses co-opt specific host lncRNAs (e.g., lncRNA-CYLD18, lncRNA-IFI661) to target and block key regulatory nodes within this pathway. By dampening the signaling cascade, such lncRNA-mediated subversion markedly attenuates IFN and ISG production, thereby facilitating viral immune evasion, efficient replication, and persistent infection. Arrow symbols are defined in the figure. All abbreviations are defined in the Glossary.
To circumvent host immune recognition, viruses co-opt lncRNAs to evade detection by PRRs. In one such mechanism, bovine viral diarrhea virus (BVDV) upregulates lncRNA lnc-CYLD, which acts as a ceRNA by sponging miR-2383 to enhance cylindromatosis lysine 63 deubiquitinase (CYLD) expression and inhibit RIG-I-mediated interferon signaling18. During pseudorabies virus (PRV) infection, the virus drives significant upregulation of host lncRNA LNC_000641. The virus-induced LNC_000641 reduces the phosphorylation levels of JAK and STAT1, and downregulates the production of IFN-α, thereby ultimately enhancing PRV replication19.
During the effector phase, functionally reprogrammed lncRNAs enable viruses to exert control over antiviral proteins. One such mechanism involves lncRNA linc-AhRA, which is dose-dependently upregulated during herpes simplex virus type 1 (HSV-1) infection via virus-induced activation of its upstream transcriptional regulator aryl hydrocarbon receptor (AhR). Functionally, linc-AhRA acts as a molecular scaffold to enhance the interaction between the E3 ubiquitin ligase TRIM27and TANK-binding kinase 1 (TBK1), thereby promoting TRIM27-catalyzed K48-linked polyubiquitination and proteasomal degradation of TBK1, and ultimately suppressing host innate antiviral immunity to facilitate viral immune evasion20. Concurrently, viruses actively suppress protective host lncRNAs to evade immune surveillance. One strategy is the downregulation of lncRNA-GM; this enhances glutathione S-transferase mu 1 (GSTM1)-mediated S-glutathionylation of TBK1, inhibiting its kinase activity and subsequent interferon production21. Through these dual strategies of hijacking and suppression, viruses construct a sophisticated immune regulatory network centered on host lncRNAs. However, the molecular mechanisms by which viruses precisely recognize and regulate such a diverse array of lncRNA molecules remain a central enigma in the field.
LncRNA-assisted viral reprogramming of the immune microenvironment
By exploiting host-derived lncRNAs, viruses not only evade innate immune responses but also modulate cellular stress and survival pathways, potentially leading to T cell and B cell exhaustion, thereby comprehensively disrupting immune homeostasis. Taking autophagy as an example, influenza A virus (IAV) significantly upregulates host lncRNA PCBP1-AS1, which encodes the proviral polypeptide PESP to enhance virus-induced autophagy via autophagy-related protein 7 (ATG7) upregulation; PESP is further stabilized by heat shock protein 90 alpha family class A member 1 (HSP90AA1) interaction, ultimately driving viral replication22. Furthermore, viruses may induce immune exhaustion through the manipulation of lncRNAs. Although direct evidence in the context of viral infection is still lacking, insights can be drawn from analogous mechanisms observed in cancer research. A relevant example is the highly expressed lncRNA Lnc-Tim3, where it specifically binds to the T-cell inhibitory receptor T-cell immunoglobulin and mucin domain-containing protein 3 (Tim-3) and competitively displaces its endogenous binding partner, HLA-B-associated transcript 3 (Bat3)23. This impairs Bat3/ lymphocyte-specific protein tyrosine kinase (Lck) complex formation, inhibiting T-cell activation, while facilitating nuclear Bat3 to enhance transcription of anti-apoptotic genes like Bcl-2, promoting survival of exhausted T cells. This provides a conceptual framework that may be applicable to viral persistence, whereby viruses could potentially exploit similar lncRNA-mediated mechanisms to drive immune exhaustion.
Fine-tuned lncRNA regulation of spatiotemporal dynamics in the viral life cycle
Viruses hijack host-derived lncRNAs to efficiently facilitate key replication steps (Fig. 5). Specific lncRNAs critically mediate viral attachment to host cell receptors. For instance, host lncRNA CCR5AS exerts an inherent proviral activity during HIV-1 infection: it stabilizes mRNA of the HIV-1 essential coreceptor C-C chemokine receptor type 5 (CCR5) by blocking RNA-binding protein RALY (RALY)-mediated mRNA degradation, thereby upregulating cell surface CCR5 expression to promote viral attachment and entry. Importantly, CCR5AS expression is not regulated by HIV-1 infection, but is strictly determined by the host rs1015164 single-nucleotide polymorphism (SNP), which modulates CCR5AS transcription by altering the binding efficiency of activating transcription factor 1 (ATF1)24. Following successful entry, viruses co-opt lncRNAs to enhance genomic replication. For example, lncRNA lnc‑ALOX12 is specifically induced in response to IAV infection; it facilitates the nuclear import of polymerase basic protein 2 (PB2) and viral RNA synthesis by binding to the IAV PB2 and sustaining its interaction with the karyopherin subunit alpha/beta 1 (importin-α/β1) complex25. In HBV infection, hepatitis B virus x protein (HBx) transcriptionally activates host lncRNA DLEU2 and directly binds to this lncRNA. The HBx-DLEU2 axis regulates the transcription of HBV cccDNA, thereby promoting viral replication26. Viruses also strategically employ specific lncRNAs to reprogram the host metabolic network. Virus-induced lncRNA-ACOD1 directly binds to the metabolic enzyme glutamic-oxaloacetic transaminase 2 (GOT2) and enhances its activity, increasing the supply of metabolites and creating an intracellular environment conducive to viral replication27. Notably, some lncRNAs possess multi-functional coordinating capabilities. LncRNA HULC is significantly upregulated upon infection with multiple hepatitis viruses, including HBV and HCV, and is hijacked by these viruses to promote viral replication through distinct regulatory pathways28,29,30. In HBV infection, HULC exerts proviral effects via two independent axes: on the one hand, it serves as a molecular scaffold to facilitate the formation of the histone acetyltransferase 1 (HAT1)/HULC/HBc complex on viral cccDNA, mediating epigenetic modifications such as histone acetylation to activate viral transcription28; on the other hand, it downregulates the restriction factor apolipoprotein B mRNA editing enzyme catalytic subunit 3B (APOBEC3B) through the HBx/STAT3/miR-539 axis, significantly enhancing cccDNA stability29. In HCV infection, HULC is induced by the viral non-structural protein 5 A (NS5A) and promotes viral replication by enhancing internal ribosome entry site (IRES)-mediated viral translation efficiency30. Collectively, these hijacked lncRNAs represent a critical virus–host interaction mechanism. By directly facilitating viral biosynthesis and reprogramming host cell status, they drive massive hijacking of cellular resources and suppression of host defense mechanisms. Given their essential roles, in-depth research into these proviral lncRNAs and the development of corresponding intervention strategies have become important directions in antiviral research.

Virally exploited lncRNAs (e.g., lncRNA-DLEU226, lncRNA-ACOD127) enhance cycle stages (entry, genome replication, assembly) and dysregulate cellular processes (e.g., lncRNA-LINC543862), enabling immune evasion, robust replication, and persistence. Arrow symbols are defined in the figure. All abbreviations are defined in the Glossary.
Intriguingly, however, a core bottleneck restricting this line of research is that the dynamic expression changes of lncRNAs during viral infection often mask fundamentally distinct regulatory logics beneath similar phenotypic patterns. Taking the downregulation of lncRNAs upon viral infection as an example, this phenotype may represent two completely different arms race strategies: host-driven active defensive downregulation, and virus-driven antagonistic downregulation. The former is exemplified by lncRNA-NRAV, which is actively downregulated by host cells upon viral infection31. As NRAV endogenously promotes viral replication by inhibiting the transcription of ISGs, its downregulation thus constitutes an effective defensive mechanism for the host to restrict viral proliferation. The latter is exemplified by lncRNA-LRIR, which serves as an intrinsic antiviral defense line by blocking influenza virus entry via suppressing transmembrane protease serine 2 (TMPRSS2) expression32. However, viruses have evolved antagonistic mechanisms to actively downregulate LRIR expression during infection, thereby relieving this blockage and sustaining their replication efficiency. These two diametrically opposed downregulation patterns vividly reveal the complex nature of lncRNAs as a central platform for host-virus arms race: an identical expression alteration can lead to completely opposite ultimate beneficiaries solely due to the difference in its driver, which also fully demonstrates the diversity of lncRNA expression and functional regulation during viral infection. The complexity of this arms race extends far beyond the functional divergence among distinct lncRNAs, and is more profoundly reflected in the fact that the same lncRNA can exert both proviral and antiviral dual roles in different contexts. This inherent functional duality renders lncRNAs veritable double-edged swords, which is precisely the core theme to be elaborated in depth in the subsequent sections.
The double-edged swords: context-driven duality of lncRNAs
Molecular mechanisms of dual-function lncRNA switching
During viral infection, multiple lncRNAs exhibit pronounced context-dependent functional duality. Specifically, rather than possessing fixed functions, a single lncRNA can exert opposing roles—proviral or antiviral—depending on the specific viral pathogen, cell type, or stage of infection. This functional plasticity is governed by a sophisticated interplay of virus-specific factors, the host cell’s metabolic, epigenetic, and immunological status, and the evolving virus–host battle. Deciphering the mechanisms behind this duality is therefore crucial for a fundamental understanding of host–virus interactions and for developing lncRNA-targeted therapies.
We discuss three representative context-dependent functional lncRNAs: NEAT1, MALAT1, and HEAL. These lncRNAs have been shown to exert opposing functions during infection by different viruses, serving as classic examples of functional duality. Their context-dependent opposing roles, associated molecular mechanisms, and key contextual determinants are summarized in Table 1, highlighting their critical regulatory function as molecular switches in virus–host interactions.
These examples demonstrate that the functions of lncRNAs are not intrinsically fixed but are shaped by their dynamic molecular environments. Key mechanisms enabling this plasticity include: (1) Transcriptional and post-transcriptional regulation: viral or host factors modulate lncRNA expression levels, splicing patterns, and modifications, thereby dynamically altering their isoform composition, stability, and function. The NEAT1 locus exemplifies this principle. It is transcribed into two overlapping isoforms, the 3.7 kb short isoform NEAT1_1 and the 23.0 kb long isoform NEAT1_2, which exhibit marked functional divergence during viral infection. During hantaan virus (HTNV) infection, NEAT1_2, but not NEAT1_1, restricts viral replication by upregulating sterol regulatory element-binding transcription factor 1 (Srebf1) and enhancing sterol regulatory element-binding protein 2 (SREBP2)-mediated inflammatory activation in macrophages, while also amplifying RIG-I-dependent type I interferon responses33. Moreover, the two isoforms are transcriptionally interdependent. Mechanistically, the essential paraspeckle protein heterogeneous nuclear ribonucleoprotein K (HNRNPK) negatively regulates NEAT1_1 3’-end polyadenylation, and this processing choice directly impacts NEAT1_2 biogenesis34. Accordingly, NEAT1_1 overexpression suppresses NEAT1_2 transcription, shifting the cellular environment toward a proviral state that exacerbates HTNV infection. This isoform-centric regulatory logic, wherein alternative 3’-end processing generates functionally opposing isoforms whose balance can be tilted by viral and cellular cues, provides a mechanistic basis for understanding how a single lncRNA locus can mount context-dependent responses to different viruses. (2) Reprogramming of interaction networks: lncRNAs selectively bind to proteins, RNAs, or DNA depending on the cellular context, enabling functional redirection through switching of molecular partners. NEAT1 exemplifies this mechanism through its virus-specific partner selection. During HTNV infection, NEAT1_2 sequesters the transcriptional repressor splicing factor proline/glutamine-rich (SFPQ), alleviating its inhibition of RIG-I and DEAD-box helicase 60 (DDX60) to amplify IFN-I responses and exert antiviral effects35. In striking contrast, during HSV-1 infection, NEAT1 recruits STAT3 via paraspeckle component 1 (PSPC1) while binding viral gene transcripts, facilitating their interaction to enhance viral gene expression36. Thus, the same lncRNA selectively exchanges its key molecular interactors depending on the infecting virus, engaging SFPQ in one context and STAT3 in another. (3) Dynamic changes in subcellular localization: their nucleocytoplasmic trafficking or aggregation into specific organelles (e.g., paraspeckles) determines the substrate molecules they interact with and their functional outcomes. The functional consequences of NEAT1’s partner switching are further shaped by the subcellular localization of these interactions. During HTNV infection, NEAT1_2 scaffolds paraspeckle assembly to sequester SFPQ away from antiviral gene promoters, thereby derepressing innate immune responses35. Conversely, during HSV-1 infection, these same paraspeckle structures are hijacked to concentrate STAT3 at viral replication sites, converting the organelle into a proviral hub36. Thus, beyond merely selecting different partners, NEAT1 directs these proteins to opposing spatial fates within paraspeckles. The sequestration of SFPQ in one context and the concentration of STAT3 in another ultimately determine whether the outcome is antiviral or proviral. These observations indicate that lncRNA subcellular localization can contribute to functional duality by spatially reorganizing their interacting partners in a context-dependent manner. (4) Cell type and infection phase specificity: the same lncRNA may exert opposite regulatory effects in different cell types or infection stages (e.g., acute vs. chronic infection) due to distinct molecular microenvironments. The opposing roles of NEAT1 during HIV-1 infection illustrate how cellular activation state dictates functional outcomes. In resting CD4⁺T cells, NEAT1 is highly expressed and exerts antiviral effects by serving as a scaffold for paraspeckle assembly, retaining unspliced HIV-1 regulator of expression of virion proteins (Rev)-dependent mRNAs that contain instability elements (INS) within the nucleus, and thereby restricting viral replication2. Upon T cell activation, however, NEAT1 expression is strongly downregulated as part of the physiological activation program37. This downregulation leads to loss of paraspeckle integrity and subsequent nuclear export of viral transcripts into the cytoplasm, creating a permissive environment for HIV-1 replication. In essence, lncRNAs act as integrative platforms that interpret contextual cues from both virus and host. Their duality is not a paradox but a reflection of their embedded position within complex cellular networks, allowing for precise, context-dependent regulation of infection outcomes.
Harnessing duality for therapeutic intervention
It is noteworthy that even for lncRNAs with well-defined, conserved intrinsic pro- or antiviral functions, divergent viral pathogens can drive diametrically opposite changes in their expression during infection. The well-characterized lncRNA MALAT1 exemplifies this paradigm, as its conserved proviral function has been validated across multiple viral infection models: during HIV-1 infection, the virus actively upregulates MALAT1 expression by recruiting the host transcription factor p50 subunit of nuclear factor κB (NF-κB) to the MALAT1 promoter38, thereby directly hijacking its intrinsic proviral activity to promote the transcription and replication of the viral genome. In stark contrast, MALAT1 expression is significantly downregulated during vesicular stomatitis virus (VSV) infection39. Existing studies suggest that this phenomenon is closely associated with host cell apoptosis induced by viral infection, and that differences in the degree of apoptosis triggered by distinct viruses may be a key driver of virus-specific changes in MALAT1 expression, while the specific regulatory pathways involved remain to be fully elucidated. Critically, this diametrically opposed expression regulation does not reflect a reversal of MALAT1’s intrinsic pro-viral function. Instead, the two opposing expression phenotypes are direct outcomes of the dynamic tug-of-war between host antiviral defense and viral immune evasion in the host–virus arms race. The divergent regulatory fate of this lncRNA may be governed by a critical expression threshold that acts as a core molecular switch: when integrated competing signals from the host and virus trigger this switch, it initiates a cascade of transcriptional and post-transcriptional events to reprogram the entire lncRNA-mediated regulatory network. Therefore, decoding the key determinants that control this threshold, and how it is modulated by different viral pathogens, is not only essential for a fundamental understanding of lncRNA regulatory biology in viral infection, but also lays a critical foundational framework for subsequent translational research.
The evolutionary conservation of such dual-functional lncRNAs suggests a complex trade-off. An lncRNA like MALAT1, which can be exploited by some viruses yet is essential for defending against others, represents a calculated risk. The host may tolerate its potential betrayal because its antiviral functions are critical for survival under other circumstances, or because its primary cellular functions in homeostasis are indispensable. This co-evolutionary arms race between host and virus has shaped lncRNAs into powerful regulatory tools—their functional output is not inherently good or bad but depends on which side, host or virus, more effectively competes for and utilizes its regulatory capacity.
This evolutionary complexity directly translates into the central challenge for therapeutic intervention: how to precisely tip the balance of these dual-functional lncRNAs in favor of the host across diverse and dynamic infection contexts. Central to this effort is precise patient stratification, which depends on diagnostic tools capable of comprehensively mapping virus–host interactions prior to intervention—including viral strain identification, replication stage, infected cell types and their states—as well as monitoring key lncRNA biomarkers and their molecular interactions. Given the dynamic functions of lncRNAs, continuous monitoring during treatment is essential for timely adjustments.
Context-specific targeting is critical to minimize off-target effects. When direct lncRNA targeting poses risks, modulating downstream effectors—such as disrupting NEAT1-mediated paraspeckle assembly—offers a safer alternative. A systems-level pharmacological approach is indispensable. Optimal strategies often combine lncRNA-targeting methods (e.g., conditional activation or effector intervention) with existing antivirals or immunomodulators to mitigate functional duality risks and achieve synergy. Furthermore, computational models integrating viral kinetics, immune dynamics, and lncRNA networks—using multi-omics and machine learning—will simulate treatment outcomes and guide optimized strategies.
The dual functionality of lncRNAs reflects their deep integration into cellular networks. Harnessing this potential demands mechanistic understanding, precision diagnostics, and dynamically responsive interventions. Acknowledging and leveraging, rather than circumventing, this complexity is essential to unlocking its therapeutic promise.
LncRNAs in viral pathogenesis and antiviral strategies
Dysregulation and mechanisms in viral disease
LncRNAs critically influence viral replication and disease progression by orchestrating immune response, apoptosis, and inflammation. This mechanism has been experimentally validated in multiple important viral diseases, indicating that dysregulation of lncRNAs is a widespread and critical molecular event in virus–host interactions. Therefore, systematically elucidating the functional positioning of lncRNAs within this network has become an important research direction for revealing viral pathogenesis and developing novel intervention strategies.
Multiple studies have revealed the clinical relevance and functional mechanisms of specific lncRNAs. In COVID-19, decreased expression of lncRNAs such as LEF1-AS1, lncCEACAM21, and HCG18 is significantly associated with disease severity and poor clinical outcomes, suggesting their potential as prognostic biomarkers40. Particularly noteworthy, the expression level of LEF1-AS1 is independently correlated with mortality risk, with an area under the receiver operating characteristic curve (AUC) value as high as 0.83 for predicting severe cases, demonstrating considerable clinical value41. Conversely, certain lncRNAs play direct roles in viral pathogenesis. The virus-induced lncRNA ARGI, particularly its type 1 diabetes (T1D) risk allele variant, adopts an altered secondary structure that enables allele-dependent binding to CCCTC-binding factor (CTCF) [35]. This interaction hyperactivates the IFN-1 response, triggering inflammation in pancreatic β cells. These findings not only highlight the central role of lncRNAs in virus–host interactions but also establish clear links between molecular mechanisms and clinical phenotypes.
However, the field still faces several major challenges. Many current findings remain correlative, and there is an urgent need to validate causality through gain- and loss-of-function experiments in relevant models. As a paradigm, HOXA-AS2, a lncRNA identified significantly upregulated by lncRNA sequencing of HBV-related clinical tissues, was further verified to exert antiviral function through systematic functional validation: lentivirus-mediated overexpression was used to achieve gain-of-function, while locked nucleic acid (LNA) GapmeR was employed for specific knockdown to realize loss-of-function, and both approaches confirmed its inhibitory effect on HBV replication10. This illustrates that many uncharacterized differentially expressed lncRNAs may harbor clear antiviral or proviral functions. Systematic functional validation of these lncRNAs will not only deepen understanding of virus–host interactions but also provide abundant host-directed targets for antiviral therapy. Moreover, given the highly context-dependent nature of lncRNA functions, systematically mapping cell type-specific lncRNA expression profiles during viral infection has become a critical frontier for future research. Finally, translating these molecular discoveries into druggable targets or highly sensitive diagnostic tools will require overcoming key technical bottlenecks such as immunogenicity, specificity, and delivery issues.
Translational challenges and future therapeutic prospects
The clinical translation of lncRNAs faces three major challenges: (1) Their functions exhibit a highly context-dependent nature: the same lncRNA may play distinctly different or even opposing roles in different cell types, stages of infection, or microenvironments (e.g., NEAT1 can either promote or suppress the replication of certain viruses). This plasticity severely limits the therapeutic window and is accompanied by unpredictable off-target effects: (2) Significant shortcomings remain in nucleic acid drug delivery systems. Achieving tissue-specific targeting, improving in vivo stability, and reducing immunogenicity represent core obstacles to efficient in vivo delivery; (3) There is a severe lack of translational evidence from preclinical to clinical stages. The absence of animal models that faithfully recapitulate human infection processes, coupled with an extreme scarcity of large-scale, multi-center clinical validation studies, greatly impedes progress. This challenge is particularly pronounced for lncRNAs, which are generally poorly conserved across species. For example, the sequence conservation rate of relevant lncRNAs between humans and mice is often less than 1%, and only a small subset retain syntenic homology—often with marked differences in expression patterns42. In light of this, preclinical studies should abandon sequence similarity-centric cross-species validation, and instead focus on syntenic positions and conserved functional motifs43. Functional verification can be further performed in combination with deep sequencing and human cell-based systems, so as to reduce extrapolation bias from rodent models and improve the reliability of clinical translation.
Addressing these challenges requires the integration of multiple cutting-edge technological strategies. At the target identification stage, single-cell RNA sequencing (scRNA-seq) and spatial transcriptomics should be employed to systematically map lncRNA expression profiles and construct regulatory networks (e.g., the LnCeCell 2.0 database) to precisely define their functional contexts and mitigate off-target risks at the source44. For functional intervention, systems like CRISPR-Cas13 enable reversible and precise editing of lncRNA sequences, offering more controllable therapeutic tools to restore beneficial functions or disrupt harmful ones45. In terms of delivery systems, lipid nanoparticles (LNPs) have emerged as a mainstream choice due to their low immunogenicity, capacity for targeted modification, and high safety profile. Additionally, exosomes, as endogenous nanocarriers, possess exceptional biocompatibility and the ability to traverse physiological barriers. They can be engineered to achieve precise targeted delivery. In pharmacokinetic studies, moving beyond traditional techniques like quantitative whole-body autoradiography (QWBA) is essential. Instead, an integrated approach should be adopted, using imaging (e.g., fluorescence in situ hybridization (FISH)) and high-resolution mass spectrometry (e.g., matrix-assisted laser desorption/ionization Fourier transform ion cyclotron resonance mass spectrometry (MALDI-FT-ICR-MS)) to elucidate the in vivo distribution, metabolism, and stability of RNA drugs across multiple levels46. Ultimately, an innovative system deeply integrating interdisciplinary expertise must be established, combining nucleic acid chemistry, biomaterials, computational biology, and clinical medicine to create a full-chain translational pathway from target identification to drug delivery and from mechanistic research to clinical validation, thereby fully enabling the clinical application of lncRNAs in antiviral therapy Box 1.
Beyond host lncRNAs, an often-overlooked but highly promising class of targets are the virus-encoded lncRNAs themselves. Compared to host lncRNAs, viral lncRNAs often possess more defined functions and a narrower interactome with host factors, making them highly specific therapeutic targets. This is exemplified by the human cytomegalovirus (HCMV)-encoded lncRNA4.9, which directly binds to and inhibits the enzymatic activity of nuclear cyclic GMP-AMP synthase (cGAS) via its 75-nt stem-loop structure, thereby mediating escape from innate immune responses47. However, their low abundance and the complex virus–host interaction environment often make them evade detection. Future efforts need to optimize and integrate protein-RNA interactome technologies (e.g., crosslinking immunoprecipitation coupled with high-throughput sequencing (CLIP-seq)), highly sensitive sequencing, and gene editing tools to comprehensively elucidate the functional mechanisms of viral lncRNAs, to pioneer novel therapeutic avenues for developing precision strike antiviral strategies.