
Mosquito production
Mosquitoes from a colony of Anopheles stephensi (line Nijmegen SDA500) were used to obtain sporozoites. Larval stages were reared in water trays at a temperature of 28 ± 1 °C and a relative humidity of 80%. Adult females were transferred to incubators with a temperature of 26 ± 0.2 °C and a relative humidity of 80%. For all the experiments, 3- to 5-day-old mosquitoes were used. The Plasmodium berghei (Pb) infected mosquitoes were maintained at 21°C at 80% relative humidity.
Experimental animals, P. berghei parasite line
Female OF1 mice and female and male C57BL/6 J mice (Charles River Laboratories, France) of 4–6 weeks old were acclimatized for one week prior to the experiment. Mice were housed between 4–5 mice per cage in ventilated cages with autoclaved aspen woodchip, fun tunnel, wood chew block and nestlets (12:12 h light–dark cycle; 21 ± 2 °C; relative humidity of 55 ± 10%). During the experiment mice were fed with commercially prepared autoclaved dry rodent diet pellets and water, both available ad libitum. All animal experiments were approved with license 11,600,202,216,547 by the Competent Authority after advice on ethical evaluation by the Animal Experiments Committee Leiden and were performed in accordance with the Experiments on Animals Act (Wod, 2014) the applicable legislation in the Netherlands in accordance with the European guidelines (EU directive no 2010/63/EU). The study was executed in a licensed establishment for experimental animals, and conducted in accordance with the ARRIVE guidelines.
Humane endpoints: the animals/body condition was thoroughly examined daily. Animals were humanely sacrificed in case the following defined endpoints are reached: visible pain (abnormal posture and/or movement), abnormal behaviour (isolation, abnormal reaction to stimuli, no food and water intake). If distress of the animals was observed by the animal caretakers, this was reported to the investigators and according to the therefore mentioned criteria, the animals were taken out of the experiment and euthanized. In all experiments no mice were euthanized before termination of the experiment and no mice died before meeting criteria for euthanasia.
The LA-GAP PbΔmei2Δlisp2 parasite (2900cl3, mutant RMgm-4937; www.pberghei.eu), which is genetically attenuated by the deletion of the meiosis inhibited 2 (mei2) and liver-specific protein 2 (lisp2) genes was used2. Feeding of Anopheles stephensi mosquitoes was performed as described previously42.
In vivo immunization of mice with Pb SPZ
The study including data acquisition was performed blinded. The study was deblinded after analysis of the data.
One day prior to immunizations, mice were randomly divided into three or four different groups (4–5 mice per group, maximum of 20 mice per experiment). On the day of immunizations, salivary glands (21–23 days post blood meal) of P. berghei LA-GAP PbΔmei2Δlisp2 (Pb GA2 SPZ) infected mosquitoes were dissected in cold RPMI 1640 glutamax (Thermo Fisher). As a control, salivary glands from the same batch of uninfected mosquitoes, hence referred to as salivary gland extract (SGE), were dissected in cold RPMI 1640 glutamax (Thermo Fisher). Immediately after dissection the glands were crushed and homogenized and the total number of Pb GA2 SPZ was counted. Intravenous immunization in the tail vein was performed after warming the mice under a heath lamp set at 35 °C to dilate the veins. For intravenous immunization, 25,000 Pb GA2 SPZ were inoculated in 200 µL RPMI 1640 glutamax (Thermo Fisher). For intradermal immunizations, the mice were anesthetized with isoflurane and 125,000 live Pb GA2 SPZ were inoculated in 20 µL RPMI 1640 glutamax (Thermo Fisher) in the left shaved upper thigh. For the SGE control, an equal amount of SGE was injected in 200 µL IV or 20 µL ID RPMI 1640 glutamax (Thermo Fisher) and for the medium control, 200 µL IV or 20 µL ID RPMI 1640 glutamax (Thermo Fisher). Second immunizations were given 7 days after the first. All injections were given between 1 and 3 pm (Fig. 6).

Experimental set-up. Overview of the timeline of mice immunizations via IV or ID. 2 days after immunizations, the parasitic liver-load was determined by IVIS. For the early immune response (myeloid activation), organs were harvested 2 days post 2nd immunization ID/IV. For the late immune response (T cell activation), cells where organs were harvested 7 days post 2nd immunization ID/IV. Cells were isolated from the organs and restimulated with GA2 SPZ, LPS, PMA/Iono or unstimulated. After 4 h of incubation, cells were stained with fluorescent antibodies and measured by flow cytometry.
Determination of parasite liver load after 1
st
and 2
nd
immunization by real-time in vivo imaging
After 44 h post-immunization the parasitic liver load was determined through bioluminescent imaging. Imaging was performed using the IVIS Lumina II Imaging System (Perkin Elmer Life Sciences, Waltham, USA)43 8 min after a subcutaneous injection with D-luciferin dissolved in PBS (100 mg/kg; Caliper Life Sciences, USA). Quantitative analysis of the bioluminescence of whole bodies was performed by measuring the luminescence signal intensity using the ROI (region of interest) settings of the Living Image® 4.4 software. Blood-stage breakthroughs were checked at 5–6 days post 1st immunization and 2nd immunization. If any parasites were discovered in the blood the mice were sacrificed and eliminated from the experiment (Fig. 6).
Organ harvesting and processing
Two days or seven days after the final immunizations, mice were anesthetized via intraperitoneal injection of 10% ketamine (Dechra Pharamceuticals, Northwich, UK) with 20 mg/mL xylazine (Alfasan, Woerden, The Netherlands). Blood was collected via retro-orbital puncture using a glass capillary tube and transferred into 1.5 mL Eppendorf tubes pre-coated with heparin, and put on ice. The blood was spun down at 4 °C, plasma was collected and stored at −80 °C. The liver and lungs were perfused by slowly injecting 20 mL cold PBS via the heart, followed by dissection of the liver, lungs and spleen after IV immunizations, and liver, lungs, spleen, skin and skin dLN after ID immunizations, and placed in sterile RPMI 1640 glutamax (Thermo Fisher) on ice. The mice were killed by exsanguination.
Livers were processed via mincing with a blade and placed in a 50 mL tube with 20 mL RPMI 1640 glutamax containing 1 mg/mL Collagenase IV (Sigma-Aldrich) and 2,000 U/mL DNase I (Sigma-Aldrich) and incubated for 45 min at 37 °C, mixing once during incubation. After incubation, tubes were placed on ice and poured through a 100-micron filter (BD) and washed with 20 mL PBS supplemented with 1% Fetal Calf Serum (FCS) and 2.5 mM ethylenediamine tetra-acetic acid (EDTA, Sigma-Aldrich). The tubes were spun down at 1,500 rpm for 10 min at 4 °C and supernatants were gently taken off, after which PBS supplemented with 1% FCS and 2.5 mM EDTA was added to the pellets and tubes were spun down at 50 g for 3 min at 4 °C. To separate the immune cells from the hepatocytes, the supernatant, which contains immune cells, was gently removed. The immune cells were spun down for 10 min at 1,600 rpm at 4 °C. The supernatant was discarded, and 3 mL sterile PBS supplemented with 0.15 M NH4Cl; 1 mM KHCO3; 0.1 mM Na2EDTA was added for 2 min to lyse red blood cells, followed by the addition of 7 mL PBS supplemented with 1% FCS and 2.5 mM EDTA (Sigma-Aldrich). The tubes were spun down for 10 min at 1,600 rpm at 4 °C. The supernatant was discarded, and the pellet was resuspended in 10 mL PBS supplemented with 0.5% bovine serum albumin (Fraction V, Roche) and 10 mM ETDA (Sigma-Aldrich) and spun down for 10 min at 1,200 rpm at 4 °C. The supernatant was removed, and 35 µL CD45 MicroBeads (Miltenyi Biotec) were added and incubated for 15 min in the fridge. After incubation, the left-over beads were washed off by adding 10 mL PBS supplemented with 0.5% bovine serum albumin (Fraction V, Roche) and 10 mM ETDA and spun down for 10 min at 1,600 rpm at 4 °C. The pellets were resuspended in 5 mL PBS supplemented with 0.5% BSA (Fraction V, Roche) and 10 mM ETDA and run through a pre-wetted LS magnetic column according to protocol (Miltenyi Biotec). The columns were washed with RPMI 1640 glutamax, 5% FCS, 0.1% β-mercaptoethanol, 100U/mL penicillin, and 100 µg/mL streptomycin (culture medium) to collect the CD45+ cells. The tubes were spun down for 10 min at 1,600 rpm at 4 °C, supernatants were taken off and resuspended in 5 mL washing medium, and viable cells were counted using trypan blue and a Bürker counting chamber.
To process lungs, the lungs were chopped into small pieces using a scalpel and transferred to a 15-mL tube. To digest the tissue 5 mL RPMI 1640 glutamax supplemented with 1 mg/mL Collagenase IV (Sigma-Aldrich), 2 µL/mL DNase (Sigma-Aldrich), and 2 µL/mL of 1M CaCL2 (Sigma-Aldrich) and incubated for 30 min at 37°C. After incubation, 10 mL cold RPMI 1640 glutamax with 5% FCS was added and poured through a 100-micron filter (BD). A 1 mL syringe was used to mash the digested tissue and washed with 20 mL cold RPMI 1640 glutamax with 5% FCS. The tubes were spun down for 10 min at 1,600 rpm at 4°C, and 2 mL sterile PBS supplemented with 0.15M NH4Cl; 1mM KHCO3; 0.1 mM Na2EDTA was added for 2 min to lyse the red blood cells. After 2 min 8 mL of washing medium was added, and tubes were spun down for 10 min at 1,600 rpm at 4°C. Pellets were resuspended in 5 mL of washing medium, and viable cells were counted using trypan blue and a Bürker counting chamber.
To process the spleen and skin-draining lymph nodes, the organs were crushed with the back of a syringe. A two-fold concentrated enzyme mix in RPMI was added, end concentration of 1 mg/mL Collagenase D (Roche) and 2000 U/mL DNase (Sigma), was added and samples were incubated for 20 min at 37°C. After incubation, the cells were poured through a 100-micron filter (BD) and washed with 10 mL cold RPMI medium. The cells were spun down for 10 min at 1600 rpm at 4°C. The supernatant was discarded, and 2 mL cold sterile PBS supplemented with 0.15M NH4Cl; 1mM KHCO3; 0.1 mM Na2EDTA was added for 2 min to lyse red blood cells. Lysis was stopped by the addition of 8 mL cold RPMI, and the pellet was resuspended in 10–20 mL cold RPMI 1640 glutamax (Thermo fisher) supplemented with 100U/mL penicillin and 100 µg/mL streptomycin (Sigma-Aldrich), 0,1% β-mercaptoethanol, and 5% FCS and counted using trypan blue and a Bürker counting chamber.
To process the skin, the skin was cut into small pieces and incubated with enzymes P, D and A, diluted in buffer L according to the manufacturer’s instructions for the whole skin dissociation kit (Miltenyi). Samples were incubated for 3 h at 37°C. After incubation, 500 µL of cold RPMI 1640 glutamax (Thermo fisher) supplemented with 100U/mL penicillin and 100 µg/mL streptomycin (Sigma-Aldrich) and 5% FCS was added. The tubes were spun according to the tumor program with the MACS dissociator (Miltenyi) and centrifuged for 10 min at 1200 rpm at 4°C. The cells were poured through a 100-micron filter (BD) and washed with 10 mL cold RPMI medium. The cells were spun down for 10 min at 1200 rpm at 4 °C, and the pellet was resuspended in 1 mL cold RPMI 1640 glutamax (Thermo fisher) supplemented with 100U/mL penicillin and 100 µg/mL streptomycin (Sigma-Aldrich), 0,1% β-mercaptoethanol, and 5% FCS. Cell counts and viability were assessed using trypan blue exclusion and a Bürker counting chamber.
Cell stimulation and analysis
A total of 200,000 cells per well were added. For specific restimulation, live Plasmodium berghei GA2 SPZ in a concentration of 25,000/mL (5,000 per 200,000 cells) in culture medium: RPMI 1640 glutamax (Thermo fisher) supplemented with 100U/mL penicillin and 100 µg/mL streptomycin (Sigma-Aldrich), 0,1% β-mercaptoethanol and 5% FCS, were added to each well and spun down for 4 min at 1,200 rpm at 4°C. For the specific restimulation, 0.1 µg/mL Phorbol 12-myristate 13-acetate (PMA) + 1 µg/mL ionomycin (Iono) (Sigma-Aldrich) in culture medium was added. To measure intracellular cytokine expression, 10 µg/mL Brefeldin A (Sigma-Aldrich) was directly added. All plates for flow cytometry were incubated for 4 h at 37 °C with 5% CO2.
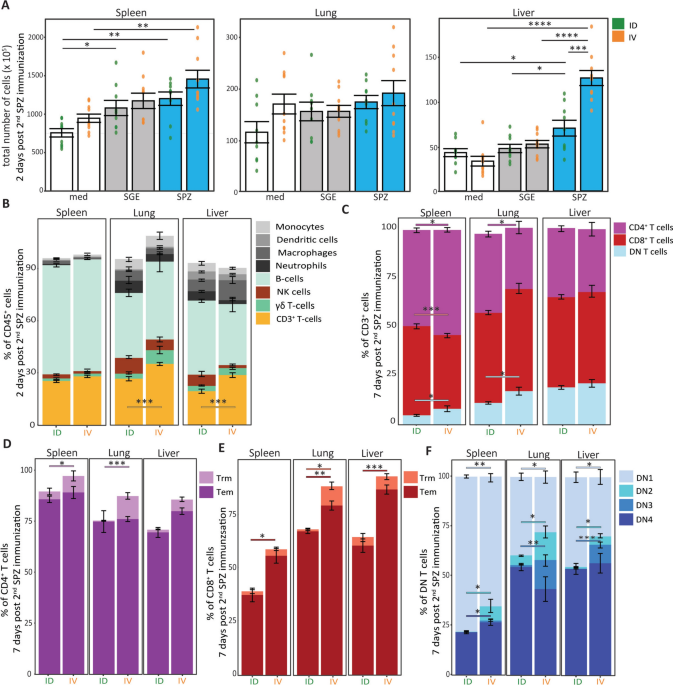
After 4 h (flow cytometry measurement), the cells were transferred into a V-bottom plate and washed with cold PBS. Cells were stained with live/dead marker Zombie NIR (Thermo Fisher), fixed with eBioscience FOXP3/Transcription Factor Fixation/Permeabilization kit, and stained with different markers for 3 different panels: (1) Myeloid panel (Table 1), (2) T-cell panel (Table 2), or (3) Advanced T-cell panel (Table 3, implemented halfway through the experiments). To all panels, Fc-block (BD bioscience), True-Stain Monocyte Blocker (BioLegend), and Brilliant Violet buffer (Thermo Fisher) was added. The cells were measured by flow cytometry using Aurora 5 laser (Cytek Bioscience B.V., Amsterdam) and analyzed using Spectroflow (Cytek Bioscience B.V.), FlowJo version 10.8 (FlowJo LLC), and R-studio version 1.4.1717. For gating strategy see supplementary Fig. 2. Of the CD4+ and CD8+ T-cells were gated into effector memory T cells (Tem, CD44hi CD62L– CD69–), resident memory -cells (Trm, CD44hi CD62L– CD69+) and DN cells into DN1, DN2, DN3 and DN4 based on expression of CD44 and CD25 expression.
Statistical analysis
Data was analysed using Spectroflow (Cytek Bioscience B.V., Amsterdam) and FlowJo version 10.8 (FlowJo LLC, Ashland, OR, USA), R studio version 4.3.1 and Adobe Illustrator 2023. Sample size estimation was performed with a power analysis using an alpha of 0.05 and a power of 90%. Statistical analyses were performed using one-way ANOVA with multiple comparisons with Bonferroni correction or an independent t-test with R studio version 4.3.1. Significance was defined as a p-value of less than 0.05. Data subjected to parametric statistical analyses had its normality confirmed beforehand.