
HMAs upregulate CD70, CD1d, and NKR ligands on AML blasts
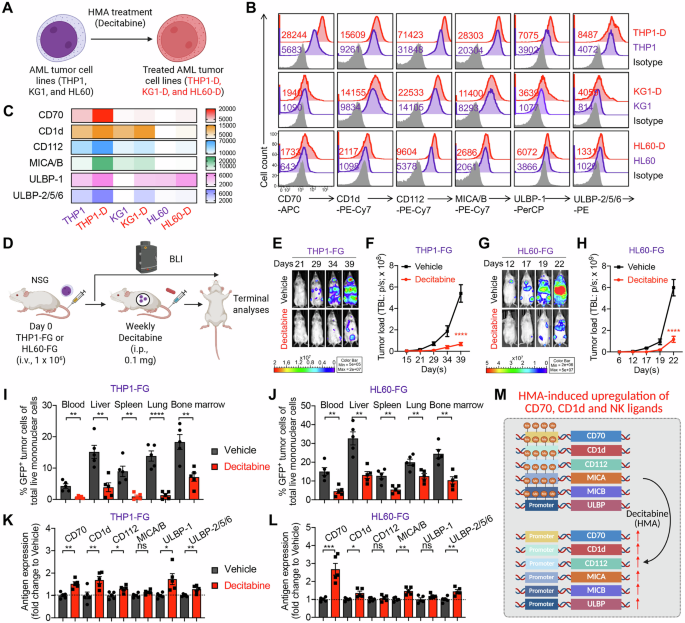
We first confirmed that HMAs upregulate the expression of CD70, CD1d, and NKR ligands on AML cells, thereby enhancing their susceptibility to CAR70-NKT cell-mediated cytotoxicity. This was validated using both in vitro cytotoxicity assays and in vivo xenograft mouse models (Fig. 1A, D). Three representative AML cell lines—THP1, KG1, and HL60—were selected to capture the heterogeneity of antigen expression observed in AML (Fig. 1A). All three expressed high levels of NKR ligands, including MICA/B and ULBP family members (NKG2D ligands), as well as CD112 and CD155 (DNAM-1 ligands) (Fig. 1B, C). Among them, THP1 cells displayed high CD70 expression, whereas KG1 and HL60 expressed minimal CD70 (Fig. 1B, C). Both THP1 and KG1 cells showed robust CD1d expression, while HL60 cells were largely CD1d-negative (Fig. 1B, C). Together, these antigenic features highlight the diversity of AML target cell phenotypes and provide a rationale for testing the combinatorial efficacy of HMAs and CAR70-NKT cell therapy across different antigen expression contexts.

A–C Antigen profiling of AML cells following HMA treatment in vitro. A Experimental design. B FACS detection of CAR target (CD70), NKT TCR target (CD1d), and NKR ligands (i.e., CD112, MICA/B, ULBP-1, and ULBP-2/5/6) on the indicated AML cells. C Heatmap showing the mean fluorescence intensity (MFI) of indicated antigen expression on AML cells. D–K Antigen profiling of AML cells following HMA treatment in vivo using THP1-FG and HL60-FG xenograft NSG mouse models. FG, firefly luciferase, and enhanced green fluorescent protein dual-reporter. E BLI images showing the presence of THP1-FG tumor cells in experimental mice over time. F Quantification of (E) (n = 5). G BLI images showing the presence of HL60-FG tumor cells in experimental mice over time. H Quantification of (G) (n = 5). I, J FACS analyses showing the percentage of tumor cells among total live mononuclear cells in the indicated tissues (n = 5). Mice inoculated with THP1-FG cells were euthanized on day 42, and mice inoculated with HL60-FG cells were euthanized on day 23. K, L FACS analyses showing the expression levels of the indicated antigens on tumor cells isolated from the bone marrow of experimental mice (n = 5). MFI values were calculated and normalized to those of tumor cells from the Vehicle group. M Diagram showing the upregulation of CD70, CD1d, and NK ligands on AML tumor cells following treatment with HMA. Representative of 2 (D–K) and 3 (A–C) experiments. Data are presented as the mean ± SEM. ns not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, by Student’s t test (I–L), or two-way ANOVA (F and H). AML acute myeloid leukemia, HMA hypomethylating agent, BLI bioluminescence imaging, i.p. intraperitoneal, FG, firefly luciferase and enhanced green fluorescent protein dual-reporter, NK natural killer.
Upon long-term, low-dose HMA (i.e., decitabine) treatment in vitro, all three AML cell lines exhibited upregulation of CD70, CD1d, and NKR ligands, albeit to varying degrees but following similar overall trends (Fig. 1B, C). Notably, HL60 cells, which originally expressed minimal CD70, showed a substantial induction of CD70 expression following HMA exposure (Fig. 1B, C), suggesting that HMA treatment may sensitize otherwise CD70-low leukemic cells to CD70-targeted therapies, including monoclonal antibodies or CD70-directed CAR approaches [23, 24].
The upregulation may result from the selective survival of cells that already express higher levels of these markers, as well as intrinsic induction of these markers following decitabine treatment. To further support this, our in vitro assays show that at a later time point, when decitabine no longer affects cell viability, the AML cell lines continue to upregulate CD70, CD1d, and NKR ligands, indicating that this enhanced expression is a direct and sustained cellular response rather than an artifact of cytotoxicity (Fig. S1A–S1C).
In addition to decitabine, we further validated our conclusions using azacitidine, another HMA commonly used to treat myeloid malignancies [10, 25]. Notably, azacitidine also significantly enhanced the expression of several tumor-associated antigens, including CD70, CD1d, and multiple NKR ligands, across all three AML cell lines (Fig. S1D and S1E). However, we observed distinct expression patterns between azacitidine and decitabine, as well as variable degrees of upregulation among the three AML cell lines. These findings indicate that HMAs can differentially modulate antigen expression in a drug- and cell line–dependent manner.
Consistent with the in vitro findings, results from in vivo xenograft models using THP1 and HL60 cells further supported the modulatory effect of HMAs (Fig. 1D). Both AML cell lines were engineered to express firefly luciferase and enhanced green fluorescent protein dual-reporter (FG), allowing longitudinal monitoring of tumor burden via bioluminescence imaging (BLI). Treatment with decitabine significantly inhibited leukemia progression, as evidenced by reduced BLI signal intensity and a lower frequency of leukemic cells detected in multiple tissues (i.e., liver, lung, and bone marrow) (Fig. 1E–J). Interestingly, AML cells isolated from HMA-treated mice exhibited enhanced expression of CD70, CD1d, and several NKR ligands (Fig. 1K, L), indicating that HMA therapy reshapes the tumor antigenic landscape and may potentiate recognition and elimination by CAR70-NKT cells (Fig. 1M).
In conclusion, these findings demonstrate that HMA treatment not only directly suppresses AML progression but also enhances tumor immunogenicity by upregulating key target and co-stimulatory ligands, thereby providing a strong mechanistic rationale for combining epigenetic therapy with CAR70-NKT cell immunotherapy.
Generate human HSPC and PBMC-derived CAR70-NKT cells
We next generated two types of CAR70-NKT cells: (1) AlloCAR70-NKT cells, derived from human cord blood HSPCs using a clinically guided culture method [21]; and (2) PBMCCAR70-NKT cells, derived from PBMCs obtained from healthy donors following a clinically adapted CAR-NKT manufacturing protocol (Fig. 2A) [26,27,28]. Because NKT cells recognize the non-polymorphic MHC class I-like molecule CD1d, they do not mediate GvHD [29,30,31,32]. Thus, both AlloCAR70-NKT and PBMCCAR70-NKT cells have the potential to serve as allogeneic, off-the-shelf cellular immunotherapies.

A Diagram showing the generation of cord blood HSPC-derived allogeneic IL-15-enhanced CAR70-NKT (AlloCAR70-NKT) cells, as well as healthy donor PBMC-derived IL-15-enhanced CAR70-NKT (PBMCCAR70-NKT) cells. B FACS plots showing the generation of AlloCAR70-NKT cells, their CD4/CD8 co-receptor profiles over the 6-week culture, and CAR70 expression on week 6 cells detected with an anti-human CD27 antibody. C FACS plots showing the generation of PBMCCAR70-NKT cells, their CD4/CD8 co-receptor profiles over the 2-week culture, and CAR70 expression on week 2 cells detected with an anti-human CD27 antibody. Purity (D), CAR expression level (E), and yield (F) of AlloCAR70-NKT and PBMCCAR70-NKT cells (n = 5; n indicated different CB or PBMC donors). G Table showing the expected yield and dose of AlloCAR70-NKT and PBMCCAR70-NKT cells when delivered to cancer patients. H ELISA measurements of human IL-15 production by AlloCAR70-NKT and PBMCCAR70-NKT cells stimulated with αGC and cultured in vitro for 5 days (n = 5). Representative of 3 experiments. Data are presented as the mean ± SEM. ns, not significant; *p < 0.05, **p < 0.01, ****p < 0.0001, by Student’s t test (D, E, and H). HSPC hematopoietic stem and progenitor cell, NKT invariant natural killer T, CAR70 CD70-targeting chimeric antigen receptor, CB cord blood, PBMC peripheral blood mononuclear cell.
For AlloCAR70-NKT cells, a lentiviral vector encoding the invariant NKT TCR (Vα24-Jα18/Vβ11), a CD70-specific CAR, and soluble IL-15 was introduced into HSPCs (Fig. 2A). The NKT TCR was cloned from healthy donor-derived NKT cells and has been previously validated to generate mature, functional NKT cells both in vitro and in vivo [19, 20, 33]. The CAR construct employs the natural CD70 ligand CD27 as its extracellular binding domain, which has been shown effective in targeting CD70-expressing malignancies such as renal cell carcinoma [18, 23, 34, 35]. Soluble IL-15 was incorporated to enhance CAR-NKT cell persistence and antitumor activity, based on extensive preclinical and clinical evidence demonstrating IL-15’s role in supporting long-term survival and function of CAR-NKT cells [36,37,38,39].
During a 6-week differentiation process, CAR-transduced HSPCs progressively generated AlloCAR70-NKT cells, increasing from approximately 20% iNKT cells at week 2 to 50% at week 4, and exceeding 97% purity by week 6 (Fig. 2B). The cells underwent typical NKT developmental progression, transitioning from CD4⁻CD8⁻ double-negative (DN) to CD4⁺CD8⁺ double-positive (DP) and ultimately to DN and CD8 single-positive (SP) populations, consistent with natural NKT cell ontogeny (Fig. 2A) [40,41,42,43].
For PBMCCAR70-NKT cells, PBMC-derived NKT cells were pre-sorted to achieve >95% purity, then activated and transduced with a lentiviral vector encoding the same CAR70 and IL-15 constructs (Fig. 2A, C). These cells retained their endogenous invariant TCR rather than receiving a transgenic iNKT TCR. After a 2-week expansion, the PBMCCAR70-NKT products maintained a stable CD4/CD8 co-receptor distribution with CD4 SP, DN, and CD8 SP subsets, suggesting that CAR transduction and expansion did not alter lineage composition (Fig. 2C) [44,45,46].
Both AlloCAR70-NKT and PBMCCAR70-NKT products achieved >97% purity, minimizing contamination by conventional αβ T cells and thereby reducing the potential risk of GvHD (Fig. 2D). However, differences in CAR expression were noted. Because endogenous NKT cells naturally express CD27, PBMCCAR70-NKT cells exhibited nearly 100% surface CD70 ligand staining, which reflects both CAR and native CD27 expression (Fig. 2C). To accurately determine the transduction rate, GFP was co-expressed from the lentivector, revealing that approximately 50–60% of PBMCCAR70-NKT cells were GFP⁺, consistent with typical lentiviral transduction efficiencies observed in PBMC-derived CAR-NKT and CAR-T cell products (Fig. 2E and Fig. S2) [18]. In contrast, AlloCAR70-NKT cells, which do not express endogenous CD27, showed uniform ( > 99%) CAR expression, likely due to the tricistronic lentivector design where CAR and NKT TCR are co-expressed (Fig. 2A) [18]. Positive selection during NKT differentiation favors dual CAR⁺/TCR⁺ cells, resulting in a homogeneous product with consistent CAR expression (Fig. 2B, E). These results indicate that the HSPC-based platform supports efficient and uniform CAR70-NKT cell generation.
Regarding scalability, from a single cord blood unit containing approximately 5 × 106 HSPCs, over 1012 AlloCAR70-NKT cells were produced, corresponding to more than 10,000 therapeutic doses based on current dosing regimens of 108–109 CAR-T cells per patient (Fig. 2F, G) [47, 48]. By comparison, one leukapheresis unit containing roughly 5 × 109 PBMCs (including ~1 × 106 NKT cells) yielded about 109 PBMCCAR70-NKT cells after 2–3 weeks of culture, sufficient for treating up to 10 patients (Fig. 2F, G). These data underscore the superior scalability and manufacturing potential of the HSPC-based allogeneic CAR-NKT platform.
Furthermore, both CAR70-NKT cell types secreted high levels of IL-15 upon stimulation with the glycolipid α-galactosylceramide (αGC), confirming functional expression and cytokine secretion capability (Fig. 2H). In conclusion, both HSPC-derived and PBMC-derived CAR70-NKT cells exhibit high purity, potent expansion capacity, and robust CAR and IL-15 expression. The HSPC-engineered AlloCAR70-NKT platform offers advantages in uniformity, scalability, and off-the-shelf potential, providing a strong foundation for future clinical translation in CD70⁺ malignancies.
Characterize human HSPC and PBMC-derived CAR70-NKT cells
We first examined CD70 expression in both AlloCAR70-NKT and PBMCCAR70-NKT cells, given prior findings in CD70-targeting CAR-T cell studies where endogenous CD70 expression on CAR-T cells induces fratricide and exhaustion, leading to reduced expansion and function [18, 35, 49]. Therefore, eliminating CD70 expression has been considered a key strategy to prevent self-targeting in CD70-directed CAR therapies. Interestingly, unlike PBMCCAR70-NKT cells which exhibited substantial CD70 expression, AlloCAR70-NKT cells did not express CD70 (Fig. 3A, B). Two additional control groups, non–CAR-engineered HSPC-derived NKT cells (AlloNKT) and PBMC-derived NKT cells (PBMCNKT), were included for comparison. Consistently, AlloNKT cells lacked CD70 expression, whereas PBMCNKT cells expressed it at high levels (Fig. 3A, B). These results suggest that the absence of CD70 expression is an intrinsic feature of HSPC-derived NKT cells, representing a natural advantage for CD70-targeting CAR-NKT therapy by preventing fratricide. Consistent with this, AlloCAR70-NKT cells exhibited robust expansion comparable to non–CAR-engineered AlloNKT cells, while PBMCCAR70-NKT cells showed markedly reduced proliferation and elevated exhaustion marker expression (Fig. 3C, D). These findings indicate that the absence of CD70 expression preserves expansion potential and prevents activation-induced dysfunction in AlloCAR70-NKT cells (Fig. 3E).

A–E Studying the fratricide risk of CAR70-NKT cells. A FACS detection of CD70 expression on AlloCAR70-NKT and PBMCCAR70-NKT cells. Non-CAR-engineered AlloNKT and PBMCNKT cells were included as controls. B Quantification of (A) (n = 5). C Fold expansion of CAR-NKT cells normalized to their non-CAR-engineered counterparts (n = 5). D Quantification of PD-1+LAG-3+TIM-3+ cells as a percentage of the total NKT cell population (n = 5). E Schematics showing the absence of surface CD70 expression on AlloCAR70-NKT cells, indicating resistance to fratricide and reduced NKT cell exhaustion. F Comparison of CD4/CD8 subpopulations of the two types of CAR70-NKT cells (n = 5). G Comparison of cytokine profiles of the two types of CAR-NKT cells (n = 5). CAR70-NKT cells were stimulated with αGC and cultured in vitro for 5 days, and the supernatants were collected for ELISA analyses. H FACS detection of intracellular IL-4 and IL-10 production by the indicated CAR70-NKT cells (n = 4). I FACS analyses of surface NK markers and NKRs on the CAR70-NKT cells (n = 4; n indicated different CB or PBMC donors). J FACS plots showing the intracellular Perforin and Granzyme B production by the indicated CAR70-NKT cells. K Quantification of (J) (n = 4; n indicated different CB or PBMC donors). Representative of 3 experiments. Data are presented as the mean ± SEM. ns, not significant; *p < 0.05, **p < 0.01, ****p < 0.0001, by Student’s t test (I and K), or one-way ANOVA (B–D, and H). SP single-positive, DP double-positive, DN double-negative, Th1 type 1 T helper, Th2 type 2 T helper, Th17 type 17 T helper, Conc. concentration, MFI mean fluorescence intensity, NKR natural killer receptor.
We next analyzed the cytokine profiles of CAR70-NKT cells. AlloCAR70-NKT cells contained predominantly DN and CD8 SP subsets, lacking the CD4 SP population commonly observed in PBMCCAR70-NKT cells (Fig. 3F). Since CD4 SP NKT cells are generally associated with helper and regulatory functions and are major producers of Th2/Th17 cytokines, their absence in AlloCAR70-NKT cells may contribute to a more cytotoxic and Th1-skewed phenotype [50,51,52]. Indeed, cytokine profiling revealed that AlloCAR70-NKT cells produced higher or comparable levels of Th1 cytokines (IFN-γ, TNF-α, and IL-2) but significantly lower levels of Th2/Th17 cytokines (IL-4, IL-10, and IL-17A), which were predominantly generated by CD4 SP PBMCCAR70-NKT cells (Fig. 3G, H). This Th1-dominant cytokine pattern aligns with a proinflammatory, antitumor phenotype favorable for tumor targeting [53,54,55,56].
Furthermore, flow cytometry analyses demonstrated that AlloCAR70-NKT cells expressed higher levels of NK-associated markers and activating receptors, including CD56, NKG2D, DNAM-1, NKp44, and NKp30, compared with PBMCCAR70-NKT cells (Fig. 3I). Correspondingly, AlloCAR70-NKT cells exhibited enhanced cytotoxic potential, evidenced by elevated expression of Perforin and Granzyme B (Fig. 3J, K). Collectively, these results indicate that AlloCAR70-NKT cells possess a less exhausted, Th1-skewed, and highly cytotoxic phenotype, making them a promising off-the-shelf cellular therapy platform for targeting CD70⁺ malignancies without the risk of fratricide.
AlloCAR70-NKT cells exhibit potent antitumor activity against AML in vitro and synergize with HMAs
We then evaluated the antitumor activity and potential synergistic mechanisms of CAR70-NKT cells in vitro using a panel of AML cell lines (THP1-FG, HL60-FG, and KG1-FG) as target cells (Fig. 3A). These AML lines represent distinct antigen-expression profiles, with or without treatment with decitabine, which upregulates CD70, CD1d, and NK ligands (Fig. 3B). Five types of effector cells were included: AlloCAR70-NKT, PBMCCAR70-NKT, non-CAR-engineered AlloNKT, non-CAR-engineered PBMCNKT, and conventional PBMC-derived T cells. Additionally, αGC was included in selected co-cultures to stimulate NKT TCR/CD1d-mediated cytotoxicity.
As expected, conventional T cells exhibited minimal tumor killing under all conditions due to the absence of CAR, TCR-mediated CD1d recognition, or NKR-mediated cytotoxicity (Fig. S3A). PBMCNKT cells did not kill AML targets without αGC, but αGC stimulation enabled killing of CD1d+ tumor cells, indicating that PBMCNKT-mediated cytotoxicity relies primarily on TCR/CD1d recognition and lacks strong NKR-mediated killing (Fig. S3A). In contrast, AlloNKT cells efficiently killed AML cells in the absence of both αGC and CD1d expression, demonstrating robust NKR-mediated tumor recognition and cytotoxicity. This mechanism was consistent with their high expression of NKRs and cytotoxic molecules and was confirmed in NKR-blocking experiments, where simultaneous blockade of NKG2D and DNAM-1 significantly reduced their antitumor activity (Fig. 3I–K and Fig. S3B).
Both AlloCAR70-NKT and PBMCCAR70-NKT cells efficiently killed CD70+ AML cells and CD1d+ targets in the presence of αGC (Fig. S4A). Notably, only AlloCAR70-NKT cells were capable of killing CD70–CD1d– AML cells via NKR-mediated recognition, highlighting a triple-targeting mechanism through CAR, TCR/CD1d, and NKRs (Fig. S3A and S3C). PBMCCAR70-NKT cells, in contrast, lacked NKR-mediated killing (Fig. S3A and S3C).
Treatment of AML cells with decitabine enhanced tumor killing by all effector cell types, reflecting increased target antigen expression (Fig. 4C). For example, PBMCCAR70-NKT cells, which were initially unable to kill HL60-FG cells without αGC, demonstrated potent cytotoxicity following decitabine-induced upregulation of CD70, CD1d, and NK ligands (Fig. 4C). Of note, AlloCAR70-NKT cells exhibited elevated IFN-γ production and Granzyme B expression when targeting decitabine-treated AML cells (Fig. 4D, E).

A Experimental design to study the synergistic effect of CAR70-NKT cells with Decitabine. B Schematics showing the indicated human AML cell lines. C Tumor cell killing data at 24 h (n = 4). AML cells were pretreated with decitabine and thoroughly washed prior to the cytotoxicity assay to ensure removal of residual drug, as decitabine is cytotoxic to therapeutic cells. D ELISA analysis of IFN-γ production by AlloCAR70-NKT cells 24 h after co-culture with the indicated AML tumor cells, in the presence or absence of αGC (n = 4). E FACS analyses of intracellular production of Granzyme B by AlloCAR70-NKT cells 24 h after co-culture with the indicated AML tumor cells, in the presence or absence of αGC (n = 4). F Diagram illustrating the upregulation of CD70, CD1d, and NK ligands on AML tumor cells treated with HMA, leading to enhanced tumor recognition and killing by AlloCAR70-NKT cells. Representative of 3 experiments. Data are presented as the mean ± SEM. ns, not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, by Student’s t test (D and E). NKL natural killer ligand, E:T effector-to-tumor cell ratio, TCR T cell receptor, αGC α-Galactosylceramide.
These results highlight a synergistic effect between CAR70-NKT cells and HMA treatment. AlloCAR70-NKT cells utilize CAR, TCR/CD1d, and NKR pathways for broad AML killing. Decitabine upregulated CD70, CD1d, and NK ligands, enhancing cytotoxicity of both AlloCAR70-NKT and PBMCCAR70-NKT cells (Fig. 4F). This combination achieves enhanced, multi-modal tumor cell killing, overcoming antigen heterogeneity and immune evasion in AML.
Lastly, we performed a side-by-side comparison of AlloCAR70-NKT cell-mediated tumor killing against AML cells pretreated with either decitabine or fludarabine. Fludarabine, a commonly used chemotherapeutic agent in AML, exerts primarily cytotoxic rather than epigenetic effects and therefore is not expected to induce substantial changes in antigen expression [57]. Consistent with this, AlloCAR70-NKT cells exhibited markedly enhanced cytotoxicity toward decitabine-treated AML cells compared with untreated controls, whereas no improvement in killing efficiency was observed following fludarabine pretreatment (Fig. S4A and S4B). These results indicate that antigen upregulation induced by HMAs, rather than general cytotoxic stress, is a key contributor to the enhanced susceptibility of AML cells to AlloCAR70-NKT cell mediated killing.
AlloCAR70-NKT cells display robust in vivo antileukemic efficacy across multiple AML models and synergize with HMAs
We evaluated the antitumor efficacy of AlloCAR70-NKT cells in vivo using two AML xenograft models: THP1-FG, representing CD70high tumor cells (Fig. 5), and HL60-FG, representing CD70low tumor cells (Fig. 6). In both models, AML cells were intravenously injected, leading to systemic dissemination to multiple organs, including liver, lung, and bone marrow, ultimately resulting in mortality. Decitabine was administered on Day 3, followed by AlloCAR70-NKT cell infusion on Day 10 to target residual or relapsed AML cells (Figs. 5A and 6A).

A Experimental design to study the synergistic effect of AlloCAR70-NKT cells with Decitabine in vivo. A human CD70+ AML cell line THP1-FG was utilized. B BLI images showing the presence of THP1-FG tumor cells in experimental mice over time. C Quantification of (B) (n = 5). D Kaplan–Meier survival curves (n = 5). E ELISA analyses of human IFN-γ and TNF-α levels in serum collected from experimental mice on day 20. F FACS analyses of the percentage of tumor cells (identified as GFP+ cells) among total live cells in bone marrow collected from experimental mice at the study endpoint. G FACS detection of AlloCAR70-NKT cells in mouse tissues on day 35. H Quantification of (G) (n = 5). I FACS detection of CD69 expression, as well as Perforin and Granzyme B production by AlloCAR70-NKT cells in mouse bone marrow on day 35. J Quantification of (I) (n = 5). Representative of 3 experiments. Data are presented as the mean ± SEM. ns, not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, by Student’s t test (H and J), one-way ANOVA (E and F), two-way ANOVA (C), or log rank (Mantel-Cox) test adjusted for multiple comparisons (D). i.v. intravenous, TBL total bioluminescence.

A Experimental design to study the synergistic effect of AlloCAR70-NKT cells with Decitabine in vivo. A human CD70– AML cell line HL60-FG was utilized. B BLI images showing the presence of HL60-FG tumor cells in experimental mice over time. C Quantification of (B) (n = 5). D Kaplan–Meier survival curves (n = 5). E ELISA analyses of human IFN-γ and TNF-α levels in serum collected from experimental mice on day 18. F FACS analyses of the percentage of AlloCAR70-NKT cells among total live mononuclear cells in mouse tissues on day 25 (n = 5). Representative of 3 experiments. Data are presented as the mean ± SEM. ns not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, by Student’s t test (F), one-way ANOVA (E), or log rank (Mantel-Cox) test adjusted for multiple comparisons (D).
In the THP1-FG xenograft model, decitabine treatment partially controlled tumor growth and extended survival; however, residual tumor cells persisted and upregulated CD70, CD1d, and NK ligands (Fig. 1E–K, and 5B–D). Subsequent AlloCAR70-NKT cell therapy achieved complete tumor elimination in all animals, resulting in long-term survival (Fig. 5B–D). This response was associated with robust in vivo effector cytokine production (IFN-γ and TNF-α) and the absence of detectable tumor cells in key tissues such as the bone marrow (Fig. 5E, F).
Analysis of AlloCAR70-NKT cell biodistribution and persistence revealed comparable frequencies in blood, spleen, and lung, with significantly higher accumulation in liver and bone marrow (Fig. 5G, H). These cells exhibited elevated activation markers (CD69) and cytotoxic molecules (Perforin and Granzyme B) while maintaining similar levels of the exhaustion marker PD-1, indicating potent and sustained effector function in target tissues (Fig. 5I, J).
Notably, AlloCAR70-NKT cells exhibited markedly superior tumor control compared with unmodified AlloNKT cells, as reflected by substantially reduced leukemic burden and prolonged survival in treated animals (Fig. S5A–S5C). These findings underscore the importance of CAR70 engineering in enhancing the antitumor potency of NKT cells, enabling more efficient recognition and elimination of CD70⁺ AML cells in vivo.
In the HL60-FG xenograft model, AlloCAR70-NKT cells demonstrated limited efficacy against CD70low tumor cells when administered alone (Fig. 6B–D). However, combination with decitabine significantly enhanced tumor control, prolonged survival, increased effector cytokine production, and promoted expansion of AlloCAR70-NKT cells in organs such as liver and lung (Fig. 6B–F). To further substantiate the requirement for HMA priming in CD70low AML, we expanded our analyses to include an additional CD70low AML cell line, KG1-FG, which exhibited similarly low basal CD70 expression as HL60-FG cells (Fig. 1B, C). Consistently, decitabine treatment induced enhanced KG1-FG susceptibility to AlloCAR70-NKT cell-mediated killing (Fig. S6A–S6D). Together, these results demonstrate that HMA priming is required in CD70low AML contexts to enable efficient AlloCAR70-NKT cell targeting and anti-leukemic activity.
Overall, these findings demonstrate that AlloCAR70-NKT cells mediate robust antitumor activity in vivo, and that synergistic treatment with HMAs enhances tumor recognition, effector function, and survival, even against antigen-low or CD70-negative AML cells.
AlloCAR70-NKT cells demonstrate a favorable safety profile in vivo
Lastly, we evaluated the safety profile of CAR70-NKT cells, focusing on GvHD, CRS, and long-term tissue toxicity, as these are major concerns for the translational and clinical development of allogeneic cell therapies [58,59,60,61]. Healthy donor PBMC-derived conventional CAR70-engineered T (CAR70-T) cells were included as a positive control (Fig. S7).
GvHD is a primary safety concern for allogeneic cell therapies [62,63,64]. We assessed xenogeneic GvHD in NSG mice by intravenously injecting three types of therapeutic cells and monitoring the development of GvHD (Fig. 7A). Conventional CAR70-T cells rapidly induced significant xenogeneic GvHD, as evidenced by weight loss, elevated clinical GvHD scores, and early mortality (Fig. 7B–D). In contrast, both AlloCAR70-NKT and PBMCCAR70-NKT cells did not induce GvHD (Fig. 7B–D). This is consistent with previous preclinical studies and clinical reports, due to their TCRs recognizing CD1d rather than polymorphic MHC, limiting alloreactivity [22, 27, 29, 30, 65, 66].

A–D Studying the GvHD risk of CAR70-NKT cells using a human xenograft NSG mouse model. Conventional CD70-targeting CAR-engineered T (CAR70-T) cells were included as a control. A Experimental design. B Clinical GvHD score recorded over time (n = 5). The score was calculated as the sum of individual scores of 6 categories (body weight, activity, posture, skin thickening, diarrhea, and dishevelment; score 0–2 for each category). C Body weight measured over time (n = 5). D Kaplan–Meier survival curves (n = 5). E–I Studying the CRS toxicity induced by CAR70-NKT cells using a THP1-FG human AML xenograft NSG mouse model. E Experimental design. F Body weight of experimental mice over time (n = 5). G ELISA analyses of mouse IL-6 and SAA-3 in mouse serum (n = 5). H ELISA analyses of human cytokines (i.e., IFN-γ, TNF-α, IL-2, and IL-6) in mouse serum (n = 5). I Quantification of mouse peritoneal macrophage numbers from the indicated samples. Mouse macrophages were identified as mouse CD45+CD11b+GR1– cells. Representative of 3 experiments. Data are presented as the mean ± SEM. ns, not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, by one-way ANOVA (G–I), two-way ANOVA (B and C), or log rank (Mantel-Cox) test adjusted for multiple comparisons (D). GvHD graft-versus-host disease, CRS cytokine release syndrome, SAA-3 serum amyloid A 3.
CRS is a common toxicity associated with CAR-T cell therapy, triggered by overactivation of immune effector cells and excessive proinflammatory cytokine release [67,68,69]. Using a high-tumor-load xenograft mouse model where tumor cells and therapeutic cells were co-injected intraperitoneally (Fig. 7E) [67], conventional CAR70-T cells caused severe CRS, evidenced by rapid body weight loss and elevated serum levels of CRS-associated biomarkers, including mouse SAA-3 and IL-6 (Fig. 7F, G). In contrast, both AlloCAR70-NKT and PBMCCAR70-NKT cells did not induce these responses, indicating a lower CRS risk (Fig. 7F, G). Notably, AlloCAR70-NKT cells produced comparable or higher levels of human Th1 cytokines (IFN-γ, TNF-α, IL-2) relative to PBMCCAR70-NKT and CAR70-T cells, but did not generate human IL-6, unlike the other cell types (Fig. 7H). The low CRS potential of CAR70-NKT cells likely reflects their NK-like features and their capacity to deplete macrophages in vivo that drive CRS (Fig. 7I) [37, 38, 55, 70].
To assess potential dose-dependent toxicity, AlloCAR70-NKT cells were administered intravenously at 1, 3, 5, or 10 × 106 cells per mouse (Fig. S8A). No acute toxicity was observed (Fig. S8B and S8C). Long-term tissue analyses conducted 120 days post-injection revealed no detectable organ toxicity or histopathological abnormalities (Fig. S8D). Overall, these studies demonstrate that AlloCAR70-NKT cells exhibit a high safety profile, with no detectable GvHD, minimal CRS risk, and no long-term tissue toxicity, supporting their translational and clinical development as a potent and safer alternative to conventional CAR-T cell therapy in the treatment of cancers including AML.