
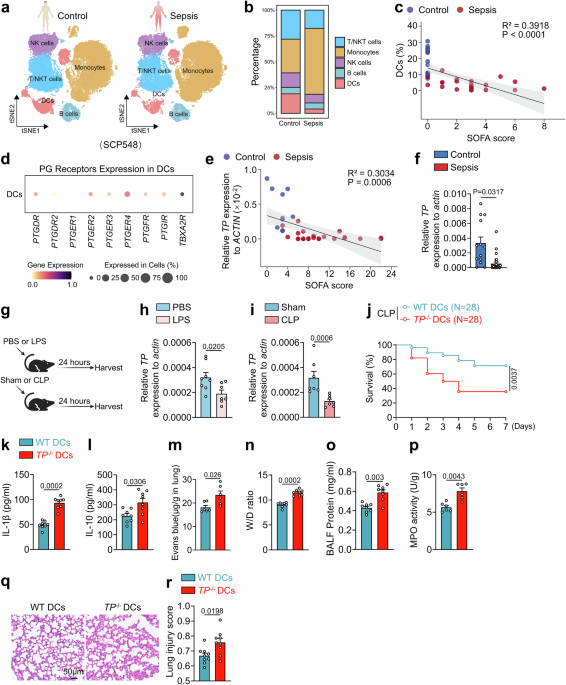
Human participants
Sepsis patients and controls were recruited at Tianjin Medical University General Hospital. Sepsis was diagnosed according to the 2016 Sepsis-3 criteria, defined as suspected or confirmed infection (evidenced by clinical signs of infection or receipt of antibiotics for > 48 h) with a sequential organ failure assessment (SOFA) score ≥ 2. All participants were recruited from the intensive care unit (ICU). The inclusion criteria for sepsis patients were (1) confirmed diagnosis of sepsis, (2) first ICU admission, and (3) aged > 18 years. The exclusion criteria were as follows: (1) received immunosuppressive therapies; (2) were pregnant or lactating; (3) had a history of malignant tumors or hematological disorders; (4) had psychiatric conditions; and (5) had incomplete clinical documentation. The inclusion criteria for the control group were as follows: (1) normal serum levels of C-reactive protein (CRP) and procalcitonin (PCT) with no fever within the previous week, and (2) aged ≥ 18 years. The exclusion criteria for controls were as follows: (1) immunosuppressive therapy; (2) body temperature > 37.3 °C at recruitment; (3) positive blood culture; and (4) abdominal surgery within the past 48 hours. Overall, 24 patients with sepsis and 13 healthy controls were enrolled in this study (Supplementary Table 1 and Table 2). This study was approved by the Institutional Ethics Committee of Tianjin Medical University General Hospital (IRB2021-YX-258-01), and written informed consent was obtained from all participants.
Cells
DC2.4 cells (Cellverse Co., Cat# iCell-m016) were maintained in our laboratory. DC2.4 cells were cultured in RPMI-1640 medium (Gibco, Cat# 11875093) supplemented with 10% fetal bovine serum (FBS) (Gibco, Cat# 10099141 C), 10 ng/mL murine granulocyte macrophage-colony stimulating factor (GM-CSF) (PeproTech, Cat# 315-03-20UG), and 100 U/mL penicillin‒streptomycin (Gibco, Cat #15140122). All the cells were incubated at 37 °C in 5% CO2.
Mice
C57BL/6 J mice (8–10 weeks) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). The TP−/−, TPflox/flox, and CD11cCre mice were bred in the laboratory. S100a9flox/flox mice were obtained from Cyagen Bioscience, Inc. (Jiangsu, China). CD11cCre mice were crossed with TPflox/flox or S100a9flox/flox mice to generate TPflox/floxCD11ccre or S100a9flox/floxCD11cCre mice. TPflox/floxS100a9flox/floxCD11cCre mice were generated by crossing TPflox/floxCD11cCre and S100a9flox/flox mice. The genetic background of all the mice was C57BL/6J. Male mice were used to avoid hormonal effects.52 The mice were maintained in an environment with a temperature of 22 °C ± 1 °C and a humidity of 50 ± 5% on a 12:12 h light/dark cycle, with free access to sterile food and water. All animal experiments were performed with the approval of the Laboratory Animal Management and Use Committee of Tianjin Medical University.
Induction of murine sepsis
The mice were assigned a numerical code and randomly assigned to either the experimental treatment group or the control group via a random table. Sepsis was induced in mice via the CLP model, which was performed as previously described.53 Briefly, the mice were anesthetized with isoflurane, the abdomen was disinfected, and a midline incision was made to expose the cecum. The cecum was ligated below the ileocecal valve and punctured with a 16-gauge needle, and a small amount of fecal content was extruded by gentle compression. The incision was closed, and the animals received 1 mL of 0.9% saline subcutaneously for fluid resuscitation. Postoperative analgesia was provided by buprenorphine (0.05 mg/kg, s.c.). Sham-operated mice underwent cecal exposure without ligation or puncture. For the alternative endotoxemia model, the mice received an intravenous injection of LPS (10 mg/kg, Sigma‒Aldrich, Cat# L2880). The animals were euthanized with CO₂ at the indicated time points for subsequent assays. To evaluate the therapeutic effect of S100a8/a9 inhibition, the mice were orally administered paquinimod (10 mg/kg/day, MCE, Cat# HY-100442) for 3 consecutive days following CLP. To assess the impact of S100a8/a9 receptor inhibition, the mice were intraperitoneally injected with either Resatorvid (3 mg/kg/day, MCE, Cat# HY-11109) or FPS-ZM1 (10 mg/kg/day, MCE, Cat# HY-19370) for 3 days post-CLP. Additionally, to investigate the therapeutic potential of TP receptor activation, the mice were intraperitoneally injected with DCpep-U-46619 (5 μg/kg/day) beginning at 2 h or 24 h after CLP induction, with continuous treatment for 3 days.
Cell isolation, culture, and transfection
PBMCs were isolated from human venous blood samples via Ficoll‒Hypaque density gradient centrifugation (Cytiva, Cat# 17144002). A single-cell suspension from a mouse spleen was obtained via mechanical dissociation through a 100 μm sieve. The DCs were purified via CD11c UltraPure microbeads (Miltenyi, Cat# 130-125-835), achieving a purity of > 85%, as determined via flow cytometry. The DCs were cultured in RPMI-1640 medium supplemented with 1% penicillin‒streptomycin and 10% FBS. Bone marrow single-cell suspensions were prepared by flushing the bone marrow through a 100 μm sieve. Bone marrow-derived neutrophils were isolated via Percoll gradient centrifugation (Solarbio, Cat# P8370) following a previously described protocol.54
DC2.4 cells were transfected with siRNA via TransIntroTM EL transfection reagent (TransGen Biotech, Cat# FT201-01). Briefly, approximately 2 × 104 DCs were seeded into 24-well plates. In a separate microfuge tube, 50 µL of Opti-MEM was mixed with 2 µL of EL transfection reagent and 50 pmol of siRNA, followed by incubation at room temperature for 20 min. The Opti-MEM-EL-siRNA mixture was then added to the cells, and the 24-well plate was gently shaken. Twenty-four hours post-transfection, the cells were harvested for mRNA expression analysis.
Adoptive transfer of DCs
To evaluate the role of TP in DCs during sepsis, WT/TP−/− DCs were isolated from mouse spleens via CD11c UltraPure microbeads. Approximately 1 × 106 DCs were injected intravenously via the caudal vein into WT recipient mice 1 h before CLP challenge.
Flow cytometry
PBMCs and single-cell suspensions from the spleen or lung were incubated at 4 °C for 30–40 min in phosphate-buffered saline (PBS) containing 1% FBS and fluorochrome-conjugated antibodies. Cell identification by flow cytometry: Human DCs: CD3⁻CD11c⁺HLA-DR⁺. Human neutrophils: CD3⁻CD11b⁺CD15⁺. Mouse DCs: CD3⁻CD11c⁺HLA-DR⁺. Mouse neutrophils: CD45⁺CD3⁻CD11b⁺Ly6G⁺. Mouse B cells: CD45⁺CD3⁻B220⁺. Mouse CD8+ T cells: CD45⁺CD3⁺CD8a⁺. Mouse T helper subsets: TH1: CD45⁺CD3⁺CD4⁺IFN-γ⁺; TH2: CD45⁺CD3⁺CD4⁺IL-4⁺; TH17: CD45⁺CD3⁺CD4⁺IL-17A⁺. Mouse regulatory T cells (Tregs): CD45⁺CD3⁺CD4⁺Foxp3⁺. Flow cytometry was performed using a BD LSRFortessa Fortessa Cell Analyzer (BD Biosciences). Data acquisition and analysis were conducted via FlowJo 8.3.3 software (TreeStar). The cells were sorted via a BD FACSAria Fusion flow cytometer (BD Biosciences).
Enzyme-linked immunosorbent assay (ELISA)
To assess cytokine levels in septic mice and cell culture supernatants, an ELISA was performed according to the manufacturer’s instructions. The cytokine kits and their detection range were as follows: IL-1β (31.3–2000 pg/mL) (DAKEWE, Cat# 1210122), IL-10 (62.5–4000 pg/mL) (DAKEWE, Cat# 1211002), S100a8 (2.7–2000 ng/mL) (CUSABIO, Cat# CSB-EL020641MO), and S100a9 (0.45–30 ng/mL) (CUSABIO, Cat# CSB-EL020642MO). The cytokine concentration in the serum was detected without dilution. Cytokine levels in the serum were expressed as absolute concentrations (pg/mL or ng/mL).
Histological analysis
The mice were euthanized, and the lungs, hearts, spleens, livers, and kidneys were harvested and fixed in 4% paraformaldehyde for 48 h. The tissues were then paraffin-embedded, sectioned at a thickness of 8 μm, and stained with hematoxylin and eosin for histological examination. Lung injury severity was quantitatively assessed via a lung injury scoring system as previously described.55
Lung vascular permeability measurements
Pulmonary vascular permeability was assessed via Evans blue–albumin (EBA) extravasation as previously described.39,56 Briefly, anesthetized mice were injected intravenously with 150 μL of EBA solution (1% Evans blue dye and 4% albumin in PBS) and allowed to circulate for 30 min. Mice were then euthanized, and the lungs were perfused with 20 mL of PBS. Lung tissues were excised, weighed, homogenized in 0.5 mL of PBS, and incubated in 1 mL of formamide at 60 °C for 24 h. The Evans blue concentration in the lung homogenate supernatants was quantified spectrophotometrically at 620 nm. The extravasated dye content was expressed as μg of Evans blue dye per g of fresh lung tissue, which was calculated against a standard curve.
Myeloperoxidase (MPO) assay
MPO activity was measured via a colorimetric assay kit (Solarbio, #BC5715) according to the manufacturer’s instructions. Briefly, lung tissues were harvested, homogenized in cold PBS, and centrifuged at 12,000 × g for 10 min at 4 °C to remove insoluble material. The supernatants were collected and assayed directly. MPO activity was quantified spectrophotometrically at 460 nm and expressed as units per gram of tissue (U/g).
Chemotaxis assay
Neutrophils were labeled with carboxyfluorescein succinimidyl ester (CFSE) (BioLegend, Cat# 423801) and resuspended at a concentration of 1 × 105 cells/100 μL in RPMI-1640 medium containing 1% FBS. CFSE-labeled neutrophils were added to the upper chamber of a 24-well transwell insert (Corning, Cat# 3421). The DCs (5 × 103 cells in 200 μL) were placed in the lower chamber. The Transwell plate was incubated at 37 °C with 5% CO2 for 2 h. After incubation, neutrophil migration was assessed by imaging via a Zeiss LSM900 confocal microscope.
Immunofluorescence
After CLP induction, the lungs were excised and embedded in optimal cutting temperature (OCT) compound (SAKURA, Cat# 4583) before being quickly frozen in liquid nitrogen. The OCT-grade tissue was sectioned at a thickness of 8 μm. The sections were fixed in cold acetone and washed with PBS. After incubation with 5% bovine saline albumin in PBS for 30 min, the samples were incubated with primary antibodies against Ly6G/Ly6C (Invitrogen, Cat# 14-5931-82) and neutrophil elastase (ABclonal, Cat# A13015) for 12 h at 4 °C, followed by washing with PBS and incubation with Alexa Fluor 594- (Invitrogen, Cat# A-21207) or Alexa Fluor 488- (Invitrogen, Cat# A11008) for 2 h in the dark at room temperature. The nuclei were labeled with 4′,6-diamidino-2-phenylindole (DAPI; Solarbio, Cat# C0060) and examined via a Zeiss LSM900 confocal microscope (Carl Zeiss).
RNA extraction and real-time polymerase chain reaction (PCR)
RNA was isolated from cells or tissues via TRIzol reagent (Invitrogen, Cat# 15596018CN) according to the manufacturer’s instructions. Purity and concentration were measured via a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). The RNA was reverse transcribed to cDNA via Hifair III 1st Strand cDNA Synthesis SuperMix for qPCR (Yeason, Cat# 11141ES10). The cDNA was amplified for 40 cycles by using Hieff UNICON® Universal Blue qPCR SYBR Green Master Mix (Yeason, Cat# 11184ES03) on a Roche LightCycler 480 Instrument II (Roche). The primers used are listed in Supplementary Table 3.
Western blotting
Cellular proteins were extracted via lysis buffer containing protease inhibitors. The protein concentration was determined via a BCA protein assay kit (Thermo Fisher Scientific, Cat# 23225). An equal amount of total protein (5 μg) was loaded into each lane to ensure consistency. Lysates containing equal amounts of protein were separated via 15% sodium dodecyl sulfate‒polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membranes (Millipore, Cat# IPVH00010). The membrane was incubated with 5% skim milk (Solarbio, Cat# D8340) for 2 h and then incubated with primary antibodies at 4 °C overnight. The following primary antibodies were used: rabbit polyclonal anti-S100a8 (Proteintech, Cat# 15792-1-AP), rabbit polyclonal anti-S100a9 (Proteintech, Cat# 26992-1-AP), rabbit monoclonal anti-Stat1 (Cell Signaling Technology, Cat# 9172), rabbit polyclonal anti-pStat1 (Cell Signaling Technology, Cat# 9167), rabbit polyclonal anti-PKCβ (ABclonal, Cat# A21241), rabbit polyclonal anti-pPKCβ (ABclonal, Cat# AP1375), rabbit polyclonal anti-PKCδ (ABclonal, Cat# A7778), rabbit polyclonal anti-pPKCδ (ABclonal, Cat# AP0776), rabbit polyclonal anti-P38 (Cell Signaling Technology, Cat# 9212S), rabbit polyclonal anti-pP38 (Cell Signaling Technology, Cat# 9216S), rabbit polyclonal anti-erk (Cell Signaling Technology, Cat# 9102S), rabbit polyclonal anti-perk (Cell Signaling Technology, Cat# 9101S), rabbit polyclonal anti-Jak1 (Cell Signaling Technology, Cat# 3332S), rabbit polyclonal anti-pJak1 (Cell Signaling Technology, Cat# 3331S), rabbit polyclonal anti-Jak2 (Cell Signaling Technology, Cat# 3230S), rabbit polyclonal anti-pJak2 (Cell Signaling Technology, Cat# 3776S), rabbit polyclonal anti-Akt (Cell Signaling Technology, Cat# 4691S), rabbit polyclonal anti-pAkt (Cell Signaling Technology, Cat# 4060S), rabbit monoclonal anti-Citrullinated Histone H3 (Cell Signaling Technology, Cat# 97272), and rabbit polyclonal anti-β-actin (Abclonal, Cat# AC026). The membranes were then incubated with a horseradish peroxidase-labeled secondary antibody (Cell Signaling Technology) in blocking buffer at room temperature for 2 h. The blots were developed via an enhanced chemiluminescence reagent (Thermo Fisher Scientific). All commercial antibodies have been validated by manufacturers or in published studies. β-actin was used as the loading control in all the experiments. The blots were scanned via a Tanon imaging system (Tanon-5200Multi). Band intensities were quantified with ImageJ software. The grayscale intensity of each target protein was normalized to that of β-actin, and normalized values for the treatment groups were expressed as fold changes relative to the mean of the control group (set to 1.0).
Preparation of the DCpep peptide derivative
The DCpep peptide derivative was synthesized via Fmoc-based solid-phase peptide synthesis with 2-chlorotriphenylmethyl chloride resin. The synthesis process involved the sequential addition of N-terminal Fmoc-protected amino acids corresponding to the target sequence. Benzotriazole-N,N,N′,N′-tetramethylurea hexafluorophosphate was used as a coupling agent to facilitate peptide bond formation with the next free amino group. After the synthesis was completed, the peptide was cleaved from the resin and deprotected with trifluoroacetic acid. To enable fluorescence tracking of the nanoagent, 4-chloro-7-nitro-2,1,3-benzoxadiazole was conjugated at the N-terminus of the peptide. The crude product obtained from the cleavage reaction was further purified via high-performance liquid chromatography to obtain the final pure DCpep peptide derivative. To assess the biological safety of DCpep-U-46619, mice received a single dose (5 μg/kg/day) and were examined on days 1 and 14 postadministration. Blood samples and major organs were collected for analysis. Hematological and serum biochemical tests were conducted to evaluate safety, and tissue sections were fixed, paraffin embedded, and subjected to H&E staining for histopathological assessment.
Transmission electron microscopy (TEM)
For TEM imaging, 10 µL of a solution containing DCpep assemblies or coassemblies of DCpep with the TP agonist U-46619 were carefully deposited onto a carbon-coated copper mesh grid. Excess liquid was removed via filter paper, and the samples were subjected to negative staining with uranyl acetate to enhance contrast and improve the visualization of the nanostructures. The prepared grids were subsequently imaged at 200 kV via a Tecnai G2 F20 transmission electron microscope, which allowed high-resolution observation of the self-assembled nanostructures.
Fluorescence spectrum analysis
To assess the fluorescence properties of DCpep-U-46619, 100 µL of DCpep-U-46619 solution was added to a 96-well plate both before and after the heating/cooling self-assembly process. The fluorescence spectra were recorded using a BioTek SynergyTM 4 Hybrid Microplate Reader at an excitation wavelength of 470 nm. This analysis was performed to monitor the structural and conformational changes in the DCpep-U-46619 assemblies.
Circular dichroism spectrum
To analyze the secondary structural composition of DCpep and its coassembled complex, DCpep-U-46619, three sample solutions were prepared: a stable DCpep solution, a self-assembled DCpep solution after the heating‒cooling process, and a coassembled DCpep-U-46619 solution. For the measurements, a special light-transmitting stone clip was used for circular dichroism chromatography. A 200 µL aliquot of the prepared solution was carefully placed onto the clip, ensuring that no bubbles or impurities were introduced. A second clip was gently attached to the edge to secure the sample. CD spectra were recorded via a MOS-450 (Biologic) spectrometer at 180–250 nm. The raw data were smoothed and processed via the DICHROWEB online analysis tool (http://dichroweb.cryst.bbk.ac.uk/html/sendform.shtml) to obtain the detailed secondary structure composition. The structural elements—including α-helices, β-sheets, β-turns, and random coils—were quantitatively determined via the standard CONTIN algorithm.
Single-cell RNA sequencing and analysis
Publicly available human scRNA-seq datasets were downloaded from the single-cell portal (SCP548). Low-quality cells were removed on the basis of the following criteria: cells expressing between 200 and 2500 genes were retained, whereas cells with > 10% mitochondrial gene expression were excluded. The data were normalized via the LogNormalize function in the Seurat (v4.4.0) package. Principal component analysis (PCA) was conducted via the RunPCA function, and batch effects were corrected via the Harmony (v1.2.0) package. The cells were clustered on the basis of the top 30 principal components (PCs) and visualized via t-SNE via the RunTSNE, FindNeighbors, and FindClusters functions in Seurat, with a resolution parameter of 0.8 applied to optimize cluster identification. Clusters were annotated by matching them with known cell-type marker genes and classifying them into five major immune cell populations.
Sorted splenic DCs from two mice per group (TPflox/flox and TPflox/floxCD11cCre) were subjected to droplet-based single-cell RNA-seq via the 10X Genomics platform. Single-cell RNA libraries were generated via a single-cell 3 v2 kit (10X Genomics) following the manufacturer’s protocol. Libraries were sequenced on an Illumina NovaSeq 6000 instrument. The raw sequencing data were processed via the Cell Ranger pipeline (v7.2.0, 10X Genomics) for alignment, barcode assignment, and UMI quantification. Low-quality cells were removed on the basis of the following criteria: retained cells expressing between 200 and 7,500 genes; cells with > 20% mitochondrial gene expression; and doublets identified and filtered via the DoubletFinder package. The data were normalized via the LogNormalize function in the Seurat (v4.4.0) package. PCA was conducted via the RunPCA function, and batch effects were corrected via the Harmony (v1.2.0) package. The cells were clustered on the basis of the top 20 PCs and visualized via t-SNE via the RunTSNE, FindNeighbors, and FindClusters functions in Seurat. A resolution parameter of 0.3 was applied to optimize distinct cluster identification. Clusters were annotated by matching them with known cell-type marker genes and classifying them into ten immune cell populations.
Differentially expressed genes (DEGs) were identified via FindMarker in Seurat software. The selection criteria for significant DEGs were as follows: adjusted p value < 0.05 and absolute log2-fold change (|log2FC|) ≥ 0.5. For functional enrichment analysis, gene ontology analysis was conducted via the enrichGO function from the clusterProfiler package (v4.6.2), with a focus on biological processes enriched in significantly upregulated genes.
RNA velocity analysis
Spliced and unspliced transcript counts for each gene in every cell were quantified via Velocyto v0.17.17 in Python. RNA velocity analysis was then performed with scVelo v0.3.2. Genes expressed in at least 20 cells were retained, and the top 2000 highly variable genes were selected via scv.pp.filter_and_normalize. Duplicate cells were removed with scv.pp.remove_duplicate_cells. The neighborhood graph was computed on the basis of 30 principal components and 30 nearest neighbors via scv.pp.moments. Cellular transcriptional dynamics were inferred with the dynamical model, and RNA velocities were estimated and projected onto the tSNE embedding. The resulting velocity vectors were used to construct the velocity graph with scv.tl.velocity_graph and to visualize transcriptional trajectories as continuous streamlines via scv.pl.velocity_embedding_stream.
Single-cell regulatory network inference and clustering (SCENIC) analysis
To predict single-cell gene regulatory networks, we performed SCENIC analysis via the SCENIC (v1.3.1) package. The raw count expression matrix was processed via GENIE3 (v1.20.0) to infer the initial coexpression networks between transcription factors (TFs) and their potential target genes. Transcription factor motifs were identified via RcisTarget (v1.18.2), which incorporates regulatory motif databases for mice (mm9-tss-centered-10 kb and mm9-500 bp-upstream) to refine the TF-target interactions. The final regulon activity scores were computed for each cell via AUCell (v1.20.2), allowing the identification of key transcription factors that drive cellular states in sepsis.
Statistical analysis
Sample size determination and power analysis were performed via G*Power software (version 3.1.9.6). All the data are expressed as the means ± standard errors of the means. The data were analyzed via Prism 8 (GraphPad Prism Software). The Mann–Whitney U test was used to compare two independent groups. Two-way analysis of variance followed by Tukey’s and Kruskal–Wallis tests and Dunn’s post hoc test were used for multiple comparisons. Survival curves were used for survival analysis. Statistical significance was set at P < 0.05. Data collection and analysis were performed blindly, without disclosing the group name to the investigator.