
Study design
This study aimed to evaluate the capacity of genome-edited B cells expressing HIV bnAbs to engraft, persist, and respond to Env vaccination in NHPs, and to model these responses in vitro using SLO cultures. Two small, exploratory adoptive cell transfer and vaccination experiments were conducted across a total of five animals. These pilot studies were not designed to be statistically powered but were intended to generate preliminary data to guide the refinement of protocols and design of larger, hypothesis-driven experiments. Investigators were not blinded during sample analysis.
Ethics statement
Indian rhesus macaques (Macaca mulatta) were housed at the Wisconsin National Primate Research Center (WNPRC). Animals were monitored twice daily by veterinarians for any signs of disease, injury, pain, distress, or psychological abnormalities and treated as recommended by the veterinarian. Involved animals were not euthanized and were returned to the colony at the end of the study. All methods were performed in accordance with relevant guidelines and regulations and were approved by the University of Wisconsin-Madison College of Letters and Sciences and the Office of the Vice Chancellor for Research Institutional Animal Care and Use Committee (IACUC protocol number G006242). The animal facilities of the WNPRC are licensed by the US Department of Agriculture and accredited by AAALAC.
Leukapheresis
Large numbers of PBMCs were collected from each animal by leukapheresis. The apheresis was performed using a Spectra Optia apheresis unit (Terumo BCT) primed with autologous blood. Animals were sedated during the apheresis procedure, and catheters were placed in the femoral vein for collection and the saphenous vein(s) to return blood to the animal. The animal’s vital signs were monitored throughout the procedure (e.g., heart and respiratory rate, ECG, oxygen saturation, blood pressure, temperature, etc.). Calcium and other electrolytes were also monitored, and calcium levels adjusted with calcium gluconate as needed to maintain baseline calcium levels. Blood was processed by Ficoll-Paque Plus (Cytiva 17144002) density gradient separation for PBMCs. PBMCs were counted and aliquots frozen in CryoStor CS10 (Stem Cell 07930).
Blood, tissue, and cell collection
Ten milli liters of blood was collected into EDTA-coated vacutainer tubes (BD Biosciences 367863) for separation into plasma and PBMCs using Ficoll-Paque Plus (Cytiva) density gradient separation. Aliquots of plasma were stored at −80 °C, and PBMCs were resuspended in CryoStor for storage of aliquots at <-150 °C. 5 ml of blood was collected into Serum Separation Tubes (SSTs) to prepare serum by centrifugation. Aliquots were stored at −80 °C for use in ELISA and pseudovirus neutralization assays. Serum was heat inactivated at 42 °C for 30 min before use in assays. Lymph node biopsies were collected from an inguinal or axillary sites four weeks after prime, and one week after each boost depending on the site of vaccination. Lymphocytes were isolated by dicing the tissue and pressing it through a 70 µM cell strainer. Cells were washed in R10 media (Gibco), counted, aliquoted and frozen in CryoStor media for storage at < -150 °C.
B cell infusions and vaccinations
(s)VRC01 targeted B cells were analyzed by flow cytometry to assess engineering efficiency, resuspended 5 ml of L-15 Leibovitz sterile tissue culture media, and autologously infused into each animal. Sedated animals were fitted with a catheter into the saphenous vein and cells were slowly infused intravenously over the course of approximately one hour while being monitored by a veterinarian. In pilot-1, 12-13 days after infusion, animals were primed and then boosted subcutaneously 10 and 25 weeks after priming with endotoxin-free recombinant protein immunogens formulated with SMNP adjuvant. MD39-ferritin [63] or eOD-GT5-60mer [74] and ZM197-4mer recombinant proteins were provided by the laboratory of Dr. William Schief and SMNP was prepared as previously described by the laboratory of Dr. Darrell Irvine [64]. In pilot-1, each dose was 100 µg of protein prepared with 750 µg of SMNP as previously described [64]. Bolus subcutaneous vaccination dosages were split in half and administered bilaterally into the quadriceps. For heterologous boost, 50 µg each of eOD-GT5-60mer and ZM197-4mer was mixed with SMNP and divided between the two vaccination sites. In pilot-2, animals were primed with an escalating dose regimen of MD39-ferritin with SMNP, as previously described [69]. Briefly, two doses of prime prepared as above were administered subcutaneously at four sites (bilaterally into quadriceps and biceps) according to the schedule given in Fig. 5. Two doses of homologous boost were administered as a bolus to these same sites 10w after prime.
Cell lines
293-T and TZM-bl cells were obtained from the American Type Culture Collection (ATCC). They were cultured in DMEM (Corning) with 10% FBS (Cell Culture Collective), 1% L-glutamine, and 1% penicillin/streptomycin (P/S) (Gibco). Expi293 cells (Gibco) were used for monoclonal antibody standard production. They were maintained in Expi293 media (Gibco) according to the manufacturer instructions. A VRC01-engineered mouse pro B cell line was used as a positive control for GT8++/MD39++ flow cytometry gating of VRC01-reprogrammed NHP B cells. This line was maintained in RPMI (Gibco) with 10% FBS, 1% L-glutamine, 1% NEAA (Gibco), 1% Na Pyruvate (Gibco), and 1% P/S. 3T3-fibroblasts expressing CD40L were provided by the laboratory of Dr. Gina Doody [75]. These cells were cultured in IMDM (Gibco) supplemented with 10% FBS (Cell Culture Collective), 1% L-glutamine, 1% non-essential amino acids (NEAA), 1% Sodium Pyruvate, 1% P/S. To irradiate, cells were detached from tissue culture flasks using 10 ml Trypsin/EDTA, then 15 ml of fresh media was added (2.5-3.5 ×107 cells per T150 flask), and cells were irradiated on ice with 50 Gy for 45 min. After irradiation, cells were washed in culture media and aliquots were frozen down in IMDM with 10% FBS and 10% DMSO for storage at -80°C.
Primary B cell isolation and culture
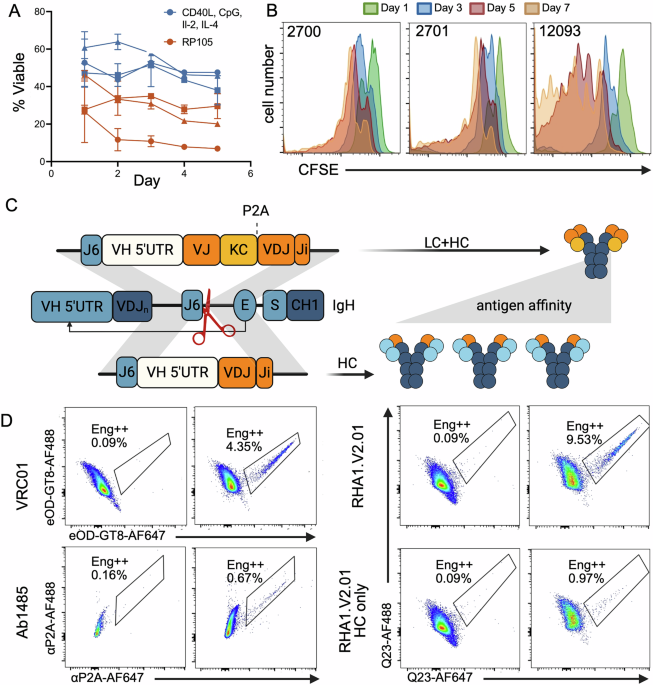
Frozen PBMCs were thawed and treated with DNase-I (StemCell, 07900) for 15 min before proceeding onto B cell isolation through negative selection with magnetic beads (StemCell, 100-0345) according to the manufacturer’s instructions. Pilot-1 and -2 culture media used the following base media: Iscove’s Modified Dulbecco’s Medium (IMDM) (Gibco), supplemented with 10% FBS (Cell Culture Collective), 1% L-glutamine, 1% MEM Non-Essential Amino Acids (NEAAs), 1% Sodium Pyruvate, 1% P/S and 0.1% ß-mercaptoethanol (BME) (Gibco). Media could be stored at 4°C in the dark for two months. Activation supplements were added directly before use. Pilot-1 activation supplements included 100 ng/mL recombinant human MEGACD40L (Enzo, ALX-522-110-C010), 2.5 µg/mL CpG ODN2006 (Adipogen, ODN2006-1), 50 ng/mL IL-2 (Peprotech, 200-02), 10 ng/mL IL-4 (Miltenyi, 130-093-922). Cells were plated at a density of 1–1.5 × 106 cells/ml and cultured for three days before changing media. To change media, cells were centrifuged at 400 g for 5 min and the pellet resuspended in fresh activation media, at a density of 1–1.5 × 106 cells/ml. Cells could be cultured this way for 9 days. Pilot-2 media included 50 ng/mL IL-4 (Miltenyi), 8 ng/ml IL-21 (Biolegend,130-093-922), and human BAFF (Biolegend, 559602). Cells were plated at a density of 0.5 × 106 cells/ml in tissue culture plates previously seeded with irradiated 3T3-human CD40L (hCD40L) feeder cells [75] at 0.5 × 106 cells/ml. After three days, cells were engineered and resuspended in fresh media and on new 3T3-hCD40-feeder cells. After 3 more days of expansion culture, cells were removed from feeder cells and cultured for a final passage at 0.5 × 106 cells/ml in media containing 100 ng/mL recombinant human MEGACD40L (Enzo) in place of feeder cells. For ImmunoCult cultures, isolated B cells were cultured using a human B cell expansion kit (StemCell, 100-0645). Cells were cultured at a density of 3–5 × 105 initially, and subsequently at 1.5 × 105 after engineering for optimal expansion. Cells were placed in fresh media every 3 days and could be cultured for up to 14 days. In all cases, cells were grown at 37 °C, with 5% CO2 and 95% humidity. To assess the mitogenic properties of activation media, 1 million B cells were stained in 5 µM of Carboxyfluorescein Succinimidyl Ester (CFSE) (Thermo, C1157) for 5 min- before placing into culture. CFSE mean florescence intensity (MFI) was assessed by flow cytometry every 24 h to monitor cell division.
Optimization of B cell nucleofection
Primary B cells were activated in culture for 24–48 h before being subjected to the T and R buffer nucleofection optimization experiments for the Neon (Invitrogen, MPK1096), according to the manufacturer’s instructions. Briefly, 0.5 M cells were washed and resuspended in 10 µl of R or T buffer containing 0.5 µg of GFP reporter mRNA (TriLink, L-8101-100). The nucleofection buffer (R/T) was never less than 80% of the total volume. Immediately after electroporation in a 10 µl Neon tip, the cells were recovered in Pilot-1 culture without antibiotics. After 2 hr, antibiotics were added back to the media. Cell viability and GFP expression was assessed by flow cytometry 24–48 h later. Electroporation conditions that balanced cell viability with high GFP expression were selected for B cell engineering.
Design and testing of IgH-targeting guide RNA
spCas9 gRNAs targeting the IgH-locus were designed using Horizon Discovery CRISPR Design Tool. Candidates with 20 nucleotide guide sequences were ordered from IDT as crRNA and complexed with trRNA (IDT) at a 1:1 mass ratio according to manufacturer instructions. Ribonucleoprotein (RNP) was prepared by complexing the crRNA/trRNA with spCas9 protein (IDT, 10007803) at a mass ratio of 1:1.2 and incubating the mixture for 20-30 min before electroporation. Electroporation Enhancer (IDT, 10007805) was added after the 20-30 min incubation, according to manufacturer instructions. After three days of culture in pilot-1 media, 3-5 × 105 cells were washed and resuspended in 10 µl R buffer containing 1 µl of RNP and electroporated with one 20 ms pulse at 1650 V in a 10 µl Neon tip. Cells were immediately recovered in activation media without antibiotics at a density of 1 × 106 cells/ml. As a control, cells were electroporated without RNP using the same conditions. Two days later, genomic DNA (gDNA) was isolated using the DNeasy kit (Qiagen). The gDNA region flanking the gRNA cut site was amplified by PCR using the following primers: 5’-AGGCCGACAGTGGTCTGGC-3’ (F) and 5’-GACACATTCC TCAGCCATCA-3’ (RC). The resulting PCR product was purified using a MinElute reaction cleanup kit (Qiagen) and Sanger-sequenced (Eton). The frequency of indels in RNP electroporated cells relative to control electroporated cells was determined using TIDE [76].
bnAb donor DNA design and production as AAV
VRC01 donor DNA was designed as follows from 5′ to 3′: (1) 554-bp HA matching sequence upstream of the Macaca mulatta IgH-J6 RNP cut-site generated by the 20 bp gRNA sequence (5’-GTCGTCGTCACCGTCTCCTC-3’), (2) rhesus IGHV4-AFI*01 gene promoter region (1500 bp 5′-UTR), (3) rhesus IGKV1-AAY*01 leader (intron removed) and mature human VRC01 kappa light-chain variable region, followed by the Macaca mulatta IGKC without stop codon, (4) GSG linker followed by a P2A [77] self-cleaving peptide sequence, (5) rhesus IGHV3-AFR*01 leader (intron removed) and mature human VRC01 heavy-chain variable region, (6) 540-bp HA downstream of the IgH-J6 RNP cut site. A splice donor consensus sequence was preserved at the VDJ-homology arm junction. The HAs and V-gene promoter were amplified from Indian rhesus macaque genomic DNA (gDNA) using the following primer sets : 1) 5’HA 5’–TGGAGGCACTTTGGAGGTCAGGAAA-3’ (F) and 5’-GAGACGGTGACGACGACCCCTTG GCCCCAGGAATCCA-3’ (RC), 2) IGHV4-AFI 5’-CCACCTGCCATATTTGCGGCAAACAATAC-3’ (F) and 5’-GTTCTTGCACAGGATGTCCGTGAC-3’ RC, 3’HA – 5’-GTAAGAATGGCC ACTCTAGAGCC-3’ F and 5’-CTCTTGGAAACCAACTTCAGGGCAC-3’ RC. Macaca mulatta HRT elements (gRNA, HAs, promoter, and leaders) were based on GenBank IGH reference MF989451.1. The VRC01 open reading frame was synthesized as a single gene (GeneArt) and all DNA fragments were assembled using Gibson assembly (NEB) into a pITR AAV backbone (provided by the laboratory of Dr. Paula Cannon) according to the manufacturer instructions and transformed into chemically competent K12 E. coli (NEB). Single colonies from bacteria plated on agar containing ampicillin were cultured. Plasmids were isolated (Qiagen) and the entire plasmid sequenced (Eton Biosciences or Plasmidsaurus). The VRC01 pITR vector was produced as the AAV6 serotype (Vector Biolabs) and stored as aliquots at −80 °C. To create HRT-AAVs with new bnAb specificities, the bnAb LC (with Macaca mulatta IGKC or IGLC*03 constant region), GSG linker, P2A peptide, bnAb HC variable region, and the first 60 bp of intronic region corresponding to the bnAb IGHJ gene (to preserve the splice donor site) was synthesized as a single gBlock (IDT) and used for cloning between the promoter and 3’ HA of the VRC01 pITR plasmid, through Gibson Assembly. Leaders corresponding to the bnAb HC and LC germline V-genes were used for each HRT design. For RHA1.V2.01 HC only design, a gBlock encoding only the bnAb HC with leader and intron region was used, omitting the bnAb LC, LC constant gene, GSG linker and P2A peptide from the design. The natural VRC01 (Genbank: GU980703.1 and GU980702.1), RHA1.V2.01 (Genbank: MT610892.1 and MT610888.1) and Ab1482 [52] variable region codons were used.
Design of simianized VRC01 bnAb donor templates
The mature VRC01 IGHV amino acid sequence was aligned to Macaca mulatta IGHV-genes [78]. 10 versions of VRC01 HC were made by manually replacing the human IGHV gene with NHP ones while conserving residues known to be important for antigen binding. A previously designed simianized VRC01 KLC [79] was paired with HC designs for expression as monoclonal NHP IgG recombinant proteins. Proteins were purified and tested in binding and neutralization assays before down selecting two variants for adaptation to HRTs based on expression levels and antigen binding properties.
B cell engineering
B cells were purified and cultured for three days in activation media as previously described. 67 h after placing cells into culture, they were washed and resuspended in serum-free IMDM at a concentration of 1×105 cells/µl and AAV6 bnAb-HRT (1-2×1012-13 genome copies (gc)/ml) (Vector Biolabs) was added at an MOI of 20,000 gc/cell. Cells were transduced with AAV for 1.5 h at 37 °C in 1.5 ml Eppendorf tubes for small volume reactions (< 3 million cells), or 50 ml conical for large volume reactions (> 3 million cells) before electroporation using the Neon nucleofection system (Invitrogen, MPK10096) to introduce CRISPR-Cas9 RNP. A 50 ml conical is used for large scale reactions to accommodate adding media for spinning down, adding enough R buffer volume and using the Neon pipette (15 ml conical is too narrow to fit Neon pipette). After transduction, 1 ml IMDM was added, and cells were spun down at 400 g for 5 min and AAV-containing supernatants were added to recovery media (activation media without antibiotics). If <1 M cells were used, spinning was omitted, and we proceeded directly with the Neon reaction. Up to 5×106 cells were then resuspended in R buffer with 10 µl RNP for nucleofection in a 100 µl neon tip using one 20 ms pulse at 1650 V. RNPs were prepared with Electroporation Enhancer as described above. R buffer was never less than 80% of the total volume in the tip. For small-scale transduction reactions of ~0.5×106 B cells, R buffer and RNP were directly added to cell pellets for electroporation in 10 µl neon tips. Cells were recovered immediately after nucleofection. Recovery media volume was always >10-fold more than the neon tip volume to sufficiently dilute the R buffer. Cells cultured in pilot-1 media were recovered at 1 ×106 cells/ml, and cells cultured in pilot-2 media were recovered at 0.5×106 until media change and expansion on day 6. Cells cultured in ImmunoCult media were recovered at a density of 3–5 × 105 for three days and subsequently cultured at 1.5×105 after engineering for optimal expansion from day 6 onward.
Engineered cell isolation
For pilot-2, engineered cells were isolated on day 6 of culture (3 days after engineering) for subsequent expansion. Feeder cells along with B cells were released from cell culture plates, with 2 µM EDTA/trypsin. Cells were then spun down at 400 g for 5 mins before proceeding with EasySep™ Release Human Biotin Positive Selection Kit (StemCell). The kit was used according to manufacturer instructions with 0.25 µg/ml of biotinylated eOD-GT8 monomer and 0.25 µl/ml of selection cocktail.
Neutralization assay
Pseudovirus production and neutralization assays were performed under sterile BSL2/3 conditions as previously described [80]. To produce pseudovirus, 293 T cells were plated with DMEM + 10% FBS + 1% P/S + 1% L-glutamine (supplemented DMEM) to achieve 50–80% confluency in 15 cm tissue culture plates (Corning, 430599) at the time of transfection. The following day, 293 T cells were co-transfected with PSG3 plasmid and pseudovirus envelope (full length) plasmid using FuGENE (Promega) and Opti-MEM transfection medium (Gibco) to produce single-round of infection-competent pseudoviruses as previously described [81]. To assess serum neutralization, 10 μl of freshly thawed, virus was diluted appropriately in supplemented DMEM and immediately mixed with 10 μl of serially diluted (2×) serum (5x starting dilution) in 384-well white plates (Corning, 3570) and incubated for 45 min at 37 °C to allow for antibody neutralization of the pseudovirus. Serum dilutions were made with supplemented DMEM and a no serum infectivity control was included. After 45 min, 3000 TZM-bl cells/well (in 20 μl of media containing 40 μg/ml Dextran) were directly added to each well containing the antibody/virus mixture. Plates were incubated at 37 °C, 5% CO2, 95% humidity for 48 h. Following the infection, media was removed and TZM-bl cells were lysed using 1× lysis buffer (25 mM Gly-Gly pH 7.8, 15 mM MgSO4, 4 mM EGTA, 1% Triton X-100) containing Bright-GloTM luciferase substrate (Promega). Virus infection of TZM-bls was assessed by luciferase intensity read on a Biotek Synergy 2 (Biotek). Infectivity data was transformed into percent virus neutralization using the equation:
$$ \% \,{neut}= \\ 100\times \frac{\left({RLU}\,{of}\,{sample}-{average}\,{RLU}\,{of}\,{cell}\,{control}\right)}{\left({average}\,{RLU}\,{of}\,{virus}\,{control}-{average}\,{RLU}\,{of}\,{cell}\,{control}\right)}$$
The serum dilution factor (SDF) values that resulted in 50% inhibition of viral infectivity (IC50) based on fitting of the % neut vs. log SDF data was calculated when possible. Functional VRC01 was quantified using the known VRC01 neutralizing IC50 for 001428_2_42 (0.019 µg/ml) which was confirmed using a VRC01-P2A-IgG standard.
Total antigen-specific IgG response enzyme-linked immunosorbent assay (ELISA)
384-well ELISA plates (Corning, 3700) were coated with 12.4 μl/well of 2 µg/ml streptavidin (JacksonImmuno #016-000-084) in PBS and incubated at 4 °C overnight. Plates were washed 3x with 100 μl/well PBS containing 0.05% Tween (PBS-T) and blocked with 60 μl/well PBS + 3% BSA at RT. After one hour, plates were washed and 12.4 μl of 2 μg/ml biotin-labeled AVI-tagged BG505 SOSIP was diluted in PBS-T and 1% BSA and added to each well. After washing (as above), rhesus sera were serially diluted (6x) using PBS-T and 1% BSA starting at 1:6 in 12.4 μl/well and incubated at RT for one hour. After washing, 12.4 μl/well alkaline phosphatase-conjugated goat anti-human IgG (H + L) (Jackson Immuno, 109-055-003) diluted 1:3000 in PBS-T, 1% BSA was added and incubated for 30 min at RT. Plates were washed and p-nitrophenyl phosphate (pNPP) substrate (Sigma Aldrich) dissolved to 1 mg/mL in substrate buffer (10 mM MgCl2 with 80 mM Na2CO3 and 15 mM NaN3, pH 9.8) was added at 12.4 μl/well to visualize the binding of antigen-specific mouse IgG. After 90 mins, optical density (OD) at 405 nm was read on an Epoch microplate spectrophotometer (Biotek). EC50 values were calculated by fitting curves to plots of Absorbance values vs. the log of the SDF for each sample using Graphpad. Variable slope (four parameter) functions with minimum (background) and maximum value constraints were used to fit curves using Prism 10 software (Graphpad). The same mass amount of biotinylated eOD-GT5 or ZM197-trimer were used instead of MD39-trimer to detect host responses to these antigens.
Anti-P2A enzyme-linked immunosorbent assays
For total P2A titer, 384-well plates were pre-coated overnight at 4 °C with 12.4 μl/well, 2 μg/ml eOD-GT8-60mer [54] in PBS. Plates were washed 3x with 100 μl/well PBS containing 0.05% Tween (PBS-T) and blocked with 60 μl/well of PBS supplemented with 3% BSA at RT for 1 h and washed again. Rhesus serum samples serially diluted (4x) with PBS-T and 1% BSA starting at 1:4 dilution were added (12.4 μl/well) and incubated at room temperature for one hour. A P2A-LC tagged human VRC01 IgG monoclonal antibody standard starting at 1 μg/mL was also added (12.4 μl/well), serially diluted (4x), and incubated at RT for one hour. Plates were washed and incubated with 12.4 μl/well biotin-labeled anti-P2A-peptide mouse antibody (Millipore Sigma, MABS2005) at 1 μg/ml in PBS-T and 1% BSA. The anti-P2A antibodies were biotinylated in-house (Thermo Scientific, PI21450). Plates were washed and incubated with 12.4 μl/well alkaline phosphatase-conjugated Streptavidin (Jackson Immuno) at 1:3000 dilution in PBS-T and 1% BSA at room temperature for one hour. Plates were washed and pNPP substrate was added, developed and read at 405 nm as above. SDF EC50 values were calculated for serum samples by fitting a curve to Absorbance vs. log SDF data points from the titration and P2A-contaning antibody concentrations in the serum could be quantified by multiplying this number to the EC50 for the standard curve. The lower limit of detection (LOD) was 4 ng/µl which corresponds to the concentration that gives an Absorbance signal that is three-times background levels. For isotype-specific P2A-titer, 384 well plates were pre-coated overnight at 4 °C with 12.4 μl/well, 2 μg/ml anti-P2A antibody (Avidity LLC, BirA500). The following day, plates were washed, blocked, and serum added as described above. Isotype-specific alkaline phosphatase-conjugated antibodies (Jackson Immuno, anti-IgG: 109-055-003, anti-IgM: 50-194-3543) were diluted to 1:1000 (anti-IgG) or 1:3000 (anti-IgM) in PBS-T and 1% BSA and added at 12.4 μl/well. Plates were washed and pNPP substrate was added, developed for 90 min before being read at 405 nm as above. P2A-titers could be quantified for IgG using a P2A-tagged IgG standard as above. IgM titers were reported as SDF EC50s. Variable slope (four parameter) functions with minimum (background) and maximum value constraints were used to fit curves using Prism 10 software (Graphpad) for unknowns and the standard. EC50 in SDF factor for unknowns were multiplied by EC50 values in µg/ml for the standard to obtain EC50s for unknowns in µg/ml.
Flow cytometry and sorting
Flow cytometry was used to detect engineered B cells using antigen-probes with specificity for reprogrammed BCRs, and for phenotyping cells cultured in activation media or SLOs as well as cells obtained from NHP PBMC or LN biopsy samples. Specific probes and protocols used for the various panels are presented in the table below. For analysis of cryopreserved samples, cells were thawed by quickly swirling cryotube in 37 °C water bath until just thawed and added to 10 ml of prewarmed supplemented IMDM media, spun at 800xg for 10 min, washed a second time with media, and then counted. Cells were then spun down in 5 ml polystyrene tubes (Falcon) and washed with PBS + 2% FBS. For live/dead staining, 1×106 cells were resuspended in 1 ml 1:1000 live/dead stain in PBS and incubated in the dark at RT for 30 mins. Cells were then washed with PBS + 2% FBS and spun again. For surface staining, 1×106 cells were resuspended per 100 µl of stain and incubated in the dark at RT for 20-30 mins (for engineered cell detection panel) or 45 mins (cell surface B cell phenotyping panel), then washed with PBS + 2% FBS and spun again. To prepare probes for detection of reprogrammed cells, biotinylated anti-P2A antibody (random biotinylation; Thermo Scientific, 21450), eOD-GT8/KO11, or native trimer probes (Q23, MD39) were conjugated with streptavidin fluorophores at appropriate molar ratios (4-Env-trimers: 1-strep-tetramer) and incubated in the dark for 15 mins before use. For intracellular staining, either True-Nuclear Transcription Factor Buffer Set (Biolegend, 424401) or Foxp3 Fix/Perm Solution (Invitrogen, 00-5523-00) were used according to manufacturer instructions. If intracellular staining was not required, cells were spun down after surface staining and resuspended in 100 µl PBS + 2% PFA and incubated at RT for 15 min, then washed and resuspended for analysis. Stained cells were resuspended in PBS + 2% FBS and acquired on a 5 L Cytek Aurora. Data was analyzed using Cytobank or FlowJo. For FACS, cells were washed once in PBS + 2% FBS then stained with probes/antibodies in PBS for 30 min at RT, if applicable. Cells were washed once in 2 PBS + 2% FBS, stained with antibodies for 45 min at 4 °C, washed twice in PBS + 2% FBS, and then resuspended in RPMI without phenol red. FACS was done on a BD AriaFusion.
|
Antibody |
Fluorophore |
Cat No |
Dilution factor or (µl/100ul stain) |
|---|---|---|---|
|
Live/dead |
Blue |
Thermo Fisher L34961 |
1:1000 (stained separately) |
|
FcX |
NA |
Biolegend 422301 |
10 |
|
BV Buffer |
NA |
BD 566385 |
10 |
|
CD3 |
BUV805 |
BD 742053 |
5 |
|
CD14 |
BUV805 |
BD 612903 |
2.5 |
|
CD20 |
BV605 |
Biolegend 302333 |
2.5 |
|
IgD |
AF488 |
Southern Biotech 2030-30 |
1.25 |
|
CD27 |
BV711 |
Biolegend 356429 |
10 |
|
CD21 |
BUV737 |
BD 612789 |
5 |
|
CD11c |
APC-R700 |
BD 566610 |
2.5 |
|
CD38 |
PerCP-Cy5.5 |
Caprico 100864 |
0.6 |
|
Lambda |
APC-vio770 |
Miltenyi 130-122-223 |
1 |
|
Bcl-6 |
BUV496 |
BD 567753 |
5 |
|
Ki67 |
BV786 |
BD 563756 |
1.25 |
|
CD80 |
PE |
BD 557227 |
5 |
|
Biotinylated-KO11 |
BV421 |
Provided by Schief Lab (Scripps) |
1 |
|
Biotinylated-eOD-GT8 |
AF555 |
Provided by Schief Lab (Scripps) |
1 |
|
Biotinylated-eOD-GT8 |
AF647 |
Provided by Schief Lab (Scripps) |
1 |
|
Biotinylated-MD39 |
AF555 |
Provided by Schief Lab (Scripps) |
2 |
|
Biotinylated-MD39 |
AF647 |
Provided by Schief Lab (Scripps) |
2 |
|
Biotinylated- Anti-P2A |
AF647 |
Millipore Sigma, MABS2005 |
1 |
|
Biotinylated- Anti-P2A |
AF555 |
Millipore Sigma, MABS2005 |
1 |
VRC01 cDNA amplification and gel
B cells were enriched from lymphocytes by MACS or specific populations were sorted by FACS. For bulk RNA libraries from sorted PBMCs, the following populations were sorted: Live CD20–IgD–CD27–CD80+CD38+, Live CD20+IgD–CD27+, LiveCD20+IgD+CD27–, and Live CD20–IgD–CD27–CD80–CD38+. Cells were sorted into RPMI with 5% FBS, centrifuged at 500xg for 10 min, then resuspended in 350 µl RLT/BME, vortexed for 1 min, and put at -80C. For bulk RNA libraries from B cells isolated from PBMCs or lymph node biopsies, B cells were isolated as described above. mRNA was purified (Qiagen), which was immediately used to generate cDNA using first-strand synthesis and random hexamers (Invitrogen) according to manufacturer instructions. Resulting cDNA was used as template in nested PCR reactions using HF-Phusion (Thermo Scientific, F530L). Primers were annealed for 30 sec at 53 °C for PCR1 and 52 °C for PCR2, with a subsequent extension time of 30 s at 72 °C. PCR products with 1X orange G were loaded onto a 1% agarose gel with 1X SybrSafe (Invitrogen) and run at 100 V in 1X TAE buffer, then imaged on a ProteinSimple gel imager.
|
PCR1 |
14 cycles |
|---|---|
|
Forward 1 |
TTCTGAAACAGGCCGGAGA |
|
IgM reverse 1 |
TACTTGCCCCCTCTCAG |
|
IgG reverse 1 |
TTGTCCACCTTGGTGTTG |
|
PCR2 |
34 cycles |
|
Forward 2 |
TGGAGGAGAATCCTGGA |
|
IgM reverse 2 |
CATTCTCACAGGAGACGAG |
|
IgG reverse 2 |
CCCTGAGGACTGTAGGA |
Recombinant antibody standards
BnAb VJ-P2A-VDJ constructs (with mouse or macaque kappa constant genes) were PCR amplified from pITR plasmids and cloned into a mouse (pFUSE-CHIg-mG1, Invivogen) or NHP IgG expression vector (pFUSE-CHIg-rhG1, Invivogen) using Gibson assembly. For mouse anti-P2A antibodies, a P2A encoding LC and HC were cloned into mouse vectors AbVec2.0-mIgkc and AbVec2.0-mIghg1, respectively (Addgene 127157, 127158). Purified (Qiagen) sequence confirmed (Plasmidsaurus) expression vectors were transfected into Expi293 cells using FectoPRO (Polyplus, 101000007) and Opti-MEM to achieve transient expression of the antibody. Expi293 cells were adjusted to a density of 2.8×106 cells/ml in 108mls of Expi293 Expression Medium. 96 µg of transfection plasmid and 96 µl of FectoPRO were mixed in 12mls total volume with pre-warmed Opti-MEM and incubated at RT for 10 min before adding to prepared Expi293 cells. Cells were fed 24 h after transfection with 1.2 mL (1% culture volume) each of 45% D-glucose and 300 mM valproic acid. Supernatants were harvested 5 days post transfection, and IgG was affinity purified using Protein G Sepharose resin (Cytiva, 17061805). Supernatants were filtered through 0.22 µm PES membranes and incubated overnight at 4 °C, rotating with 1 ml Protein G resin. The following day, supernatants were added to chromatography columns for gravity-flow purification. Columns were washed with ten column volumes of PBS. IgG was eluted with 0.2 M citric acid pH 3.0 into 20% elution volume 2 M Tris Base pH 9.0, then immediately buffer exchanged into PBS with 50 K MWCO Amicon tubes (Millipore Sigma, UFC905008).
Surface plasmon resonance
Kinetics of antibody-antigen interactions were measured on a Biacore 8 K (Cytiva). Experiments were performed at 25 °C at a flow rate of 30 µL/min in a mobile phase of HBS-EP+ (Teknova). Anti-human Fc IgG antibody was immobilized on a CM3 chip (Cytiva, BR100541) using the Human Antibody Capture Kit (Cytiva, BR100839) according to manufacturer instructions. bnAbs expressed as rhesus IgG were injected over the immobilized CM3 chip, followed by a wait period for normalization of response units (RU). Regeneration was accomplished using 3 M MgCl2 with 30 s contact time. A concentration of MD39-trimer ranging from 15.625–500 nM was injected across the antibody and control surface for 120 s, followed by a 500 s dissociation phase in PBS buffer alone. MD39-trimer binding on (kon) and off (koff) rates were assessed and KD were calculated using BIAEvaluation software (Cytiva).
Single Cell RNA sequencing and analysis of culture B cells
Rhesus macaque single-cell cDNA libraries were prepared using the 10x Genomics Chromium Next GEM Single Cell 5’ Kit (V2 chemistry) and the 10x Genomics Chromium Next GEM-X Single Cell 5’ Kit (V3 chemistry). The cDNA libraries were used to generate and GEX libraries according to the manufacturer instructions. The libraries were quantified using Agilent Tapestation and sequenced with Illumina NextSeq 1000/2000 kits. Fastq files were generated from raw sequencing base call (BCL) data using CellRanger (8.0.1) mkfastq. The fastqs from the 5’ GEX libraries generated from the cell culture samples were processed using Cellranger count with a custom Rhesus macaque reference. The reference was sourced from Ensembl and updated to include VRC01 HC and LC IGV-gene sequences, and unannotated genes were removed. Ig constant genes that were unannotated in the reference (IgD, IgA, and IgG1) were annotated using pantherdb [82]. Scanpy [83] was used to filter cells out if they expressed fewer than 100 genes and genes were discarded if they were present in fewer than three cells. Mitochondrial genes were assessed to determine quality of cells and dataset [84]. A principal component analysis was executed to reduce the dimensionality of the gene counts matrix, then the cells were Leiden clustered [85] and neighbors were embedded using UMAP [86]. Ig variable genes were removed from the dataset prior to clustering and embedding. Engineered cells were identified as positive for VRC01 IGV-gene expression. Endogenous IGHV-gene transcription could be assessed in subsets of unengineered cells when they expressed IGHV-genes that could align with greater than 90% identity (Scanpy) to several annotated IGHV-genes in the reference sequence. Isotype was determined based on raw expression values of IgH constant genes reads above an expression threshold of 250 reads/cell.
Organoid culture and vaccination
Organoids were cultured as previously described [70, 71]. Cryopreserved spleen or LN biopsied cells were cultured in 96-well transwell plates (Corning, 07200278) in RPMI with Glutamax (Gibco, 35050061), 10% FBS, 1× nonessential amino acids, 1× sodium pyruvate, 1× penicillin–streptomycin, 1× Normocin (InvivoGen, ant-nr-2) and 1× insulin/selenium/transferrin cocktail (Gibco), 1 μg/ml of recombinant human BAFF (BioLegend, 559602) at 1.5 × 106 cells/well. 30% of media was changed from the base well every other day beginning on day 1 of culture (24 h after cultures began). For vaccinations, Adju-Phos (InvivoGen, vac-phos-250) and Alhydrogel (InvivoGen, vac-alu-250) were combined with immunogen according to manufacturer instructions. For imiquimod (InvivoGen, vac-imq) and SMNP, 0.05 µg per transwell were used for each vaccination. Vaccinations were added directly to the transwell on the first day of culture. Engineered cells were vaccinated with 1 µg/ml MD39-ferritin and SMNP. Samples were assessed by flow cytometry using B cell phenotyping panel described above. Cells were removed from transwells by pipetting up and down several times, and transwells were washed with PBS with 2% FBS to remove residual cells before spinning down and proceeding to flow cytometry staining.