
{kind=link}
![[Düzenle]](?edit=/home/u514654129/domains/awesamohealth.com/public_html/wp-content/themes/pixwell/screenshot.png){kind=link}
![[Yeniden Adlandır]](?rename=/home/u514654129/domains/awesamohealth.com/public_html/wp-content/themes/pixwell/screenshot.png){kind=link}
Most mouse models recapitulate the composition of desertic, macrophage-rich human tumors
To benchmark the immune composition and transcriptomic patterns of typical murine TMEs against human TMEs, we profiled 15 widely used mouse models representing more than 95% of published immunotherapy studies. This selection includes common cell lines (notably B16F10, MC38, CT26, LLC, 4T1 or RENCA), autochthonous/transplanted (KPC) and genetically engineered models (MMTV-PyMT) across BALB/c and C57BL/6 backgrounds18,19,20,23,38 (Fig. 1a, Table 1 and Supplementary Table 1). Tumors were analyzed at day 14 after implantation (or ~500 mm3) using cytometry by time-of-flight (CyTOF) in all models and using single-cell RNA sequencing (scRNA-seq) in nine models.

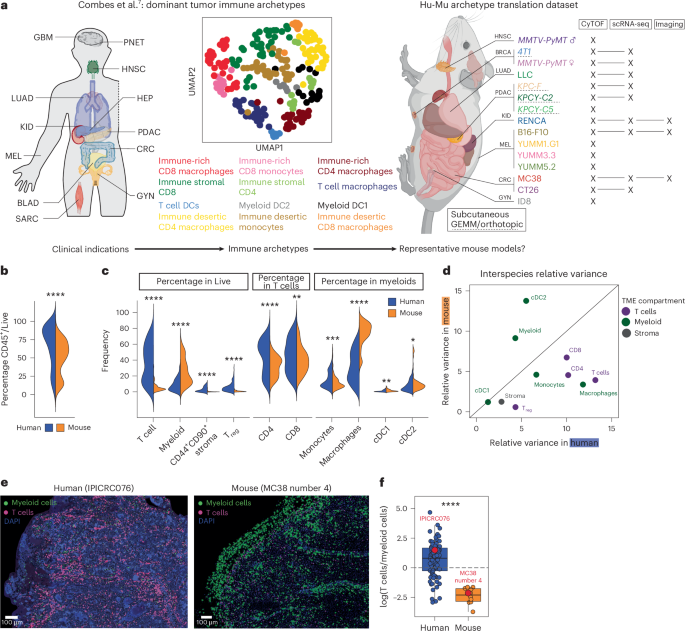
a, Schematic of our human7 and murine study cohorts. b, Violin plot presenting the frequency of CD45+ in Live cells from human (blue, all samples grouped, n = 170) versus murine (orange, all samples grouped, n = 109) tumors (P = 1.39 × 10−7). c, Violin plot presenting the frequencies of conventional T cells (P = 4.83 × 10−55), non-granulocytic myeloid cells (combining monocytes, macrophages, conventional DCs and plasmacytoid DCs, P = 2.57 × 10−19), nonimmune stroma (CD44+CD90+ in CD45−, P = 2.17 × 10−30) and CD4+ Treg cells (P = 1.65 × 10−35) out of Live cells; CD4 T conventional (P = 4.23 × 10−8) and CD8 (P = 7.31 × 10−4) out of T cells; monocytes (P = 9.55 × 10−4), macrophages (P = 1.72 × 10−47), cDC1 (P = 5.85 × 10−3) and cDC2 (P = 4.11 × 10−2) out of myeloid cells between human (blue, all samples grouped, n = 224) and mouse (orange, all samples grouped, n = 109) tumors. d, Plot comparing the relative variance of each parameter shown in b (colored according to cellular compartment) between human and murine tumors. The diagonal line represents an equal relative variance between the two species. e, Representative images showing our identification of T (pink, identified as CD3+CD4+/−CD8+/− in human and CD3+CD11b− in mouse) and myeloid (green, identified as CD3−HLA-DR+XCR1+/−CD163+/− in human and CD3−CD11b+ or CD3−MHC-II+ in mice) cells in a human (left) or murine (right) tumor slice. f, Box plot presenting the ratio of T cells over myeloid cells in human (blue, n = 85) versus murine (orange, n = 10) tumors (P = 2.1 × 10−6), calculated using a measurement based on the imaging shown in e. Boxes represent the 25th–75th percentile; the horizontal line represents the median; the whiskers represent 1.5 times the interquartile range (Tukey); and points represent individual samples. Statistical significance in all panels was calculated using a t-test with Bonferroni correction, *Padj ≤ 0.05, **Padj ≤ 0.01, ***Padj ≤ 0.001, ****Padj ≤ 0.0001. All statistical tests were two-sided; P values are reported as exact values unless otherwise indicated. GBM, glioblastoma; PNET, primitive neuroectodermal tumor; HNSC, head and neck squamous cell carcinoma; LUAD, lung adenocarcinoma; HEP, hepatic tumor; KID, kidney tumor; PDAC, pancreatic ductal adenocarcinoma; MEL, melanoma; CRC, colorectal cancer; BLAD, bladder cancer; GYN, gynecologic tumor; SARC, sarcoma; UMAP, uniform manifold approximation and projection; Hu, human; Mu, murine; BRCA, breast carcinoma; GEMM, genetically engineered mouse model; cDC, conventional dendritic cells. Panel a created in BioRender; Lab, C. https://biorender.com/vf8ffoa (2026).
Source data
As a primary comparator, we used the human ImmunoProfiler dataset, which defines immune archetypes based on human TME composition and deep transcriptomic profiling of T cell, non-granulocytic myeloid, stromal and tumor populations, and has already benchmarked multiple studies7,31,39,40 (Supplementary Table 1). By analyzing total immune frequencies across human and murine TMEs (Extended Data Fig. 1a), we found that both species displayed bimodal distributions of total immune density (‘rich’ and ‘poor’), but murine TMEs had significantly lower overall immune cell frequencies than human TMEs (Fig. 1b).
Based on our studies and others that classified human TMEs4,6,7, we used ten major cellular TME compartments to compare the composition of overall human and murine TMEs (Extended Data Fig. 1b). We found that murine TMEs consistently contained fewer T cells and higher myeloid frequencies than human TMEs, with a particular bias toward macrophage abundance (Fig. 1c). Variance across murine TMEs was driven largely by myeloid density and composition, whereas human TMEs exhibited broader T cell variability (Fig. 1d). Imaging of intact human and murine tumors confirmed these findings, showing that human tumors generally were T-cell-dominated, while mouse tumors were strongly myeloid-biased (Fig. 1e,f and Extended Data Fig. 1j).
Despite model-specific variations in murine TME composition (Extended Data Fig. 1c,d, in line with previous reports18,19,20,23) embedding mouse and human samples revealed that nearly all mouse models (except RENCA) clustered with immune-desert, macrophage-rich human archetypes (Fig. 2a,b). These two archetypes represent only ~17% of patients in our ImmunoProfiler cohort7, with indication-specific variations ranging from 0% in hepatocellular carcinomas to 46.5% in gynecological tumors (Extended Data Fig. 1e).

a, Embeddings of the 10 feature frequencies in Fig. 1c showing human samples (circles, individual samples colored according to archetype, n = 224) and mouse tumors (triangles, initial n = 109 averaged according to tumor line resulting in n = 12 data points on the graph) in the same UMAP space (left) or in a hierarchically clustered heatmap (right). For both graphs, the 10 feature frequencies were z-scored across the entire dataset (224 human + 109 mouse samples) before averaging by human archetype/mouse model and UMAP embedding/plotting. b, Hierarchically clustered matrix of cosine similarities calculated between each mouse tumor model (orange) and each human archetype (blue), using the frequencies in Fig. 1c. c, Scatter plot presenting the mapping of 2,543 mouse tumor samples from the NCBI Sequence Read Archive in the 6 immune subtypes described in ref. 3. The graph shows the percentage of mouse samples mapping to each subtype (x axis, values also displayed on the graph) and the average percentage of leukocytes infiltrated in each subtype (y axis).
Source data
To extend this analysis, we queried 2,543 additional mouse RNA-seq samples from 178 studies deposited in the NCBI’s Sequence Read Archive that we benchmarked against an established human TME classification3 built on the TCGA41. Across this large mouse cohort, more than 60% were classified as ‘lymphocyte-depleted’ (C4), while very few were classified as ‘immune-rich’ (~7% in the C2 and C6 subtypes) (Fig. 2c, Extended Data Fig. 1h,i and Supplementary Table 1). Therefore, low T cell infiltration is a dominant feature of murine tumors.
Importantly, the T cell to myeloid imbalance in murine TME (Extended Data Fig. 1f) persisted across many experimental conditions, including KPC pancreatic tumors implanted either orthotopically or subcutaneously, and in B16, LLC and MC38 tumors inoculated to either mice housed under ‘dirty’ conditions, which can promote T cell activation and accumulation in tissues42, or to aged mice or mice fed a high-fat diet (Extended Data Fig. 1g).
Taken together, these findings suggest that even though murine TMEs display model-specific variations, they predominantly resemble a minority subset of immune-desertic, macrophage-rich human TMEs.
Divergence in chemokine networks
Chemokine networks being major contributors to immune cell densities in tissues43, we performed a systematic analysis of chemokine and receptor transcript expression7,43,44 within key cell populations of the TMEs. For this, we analyzed bulk RNA-seq data from sorted T, Treg, myeloid, tumor and stroma compartments from human TMEs7, compared to pseudobulked scRNA-seq datasets from our murine TME cohort (Extended Data Fig. 2a). The mouse scRNA-seq also allowed to describe subtype-specific expression of each chemokine (Extended Data Figs. 2b–f and 3b–d) in T cells, dendritic cells, monocytes and macrophages, and nonimmune cells subclusters.
We first analyzed the expression patterns of chemokine receptors across compartment and species (Extended Data Fig. 3a–c), before focusing on T-cell-specific chemokine receptors. While several chemokine networks were conserved (CCR4, CCR7, CXCR4, CXCR6 expression patterns, as well as their ligands), key differences emerged. Notably, CCR2 and CCR5 transcripts were reduced in mouse T cells relative to human T cells (Fig. 3a,b), which was consistent across models and subsets (Extended Data Fig. 3b) and confirmed at the protein level for CCR5 (Fig. 3c).

a, Heatmap comparing the scaled expression of chemokine receptors between human and murine T cells. For each species, the expression of each receptor was extracted for all T cells (bulk-sorted in human versus pseudobulked from scRNA-seq in mice) and scaled compared with their expression in the other available TME compartments of the same species (that is, Treg, myeloid, tumor and stroma). We then extracted the scaled values for T cells and plotted them side-by-side in a heatmap. The full heatmap can be found in Extended Data Fig. 3. b, Box plots showing the expression levels of CCR2 and CCR5 across T cells, Treg, myeloid, tumor and stroma in human (top, shown as transcripts per million (TPM) from bulk RNA-seq, each dot representing a single patient) versus murine (bottom, shown as the expression level from scRNA-seq averaged per sample, each dot representing a single mouse sample) tumors, exemplifying the differences found in a. c, Flow cytometry validation of the protein expression of CCR5 in human (top) and murine (bottom) tumors across different cell compartments, plotted as representative histograms (left) and box plots of mean fluorescence intensity (MFI) (right). For the mouse box plot, T cells versus myeloid comparisons yielded P = 0.013 in B16 and P = 0.0033 in MC38; Treg versus myeloid comparisons yielded P = 0.012 in B16 and P = 0.002 in MC38. d, Heatmap (as in a) comparing the expression of specific chemokine ligands binding the receptors found conserved in a between the different cellular compartments of human versus murine tumors. The full heatmap can be found in Extended Data Fig. 3. e, Box plots as in b showing the expression levels of CCL22, CXCL9 and CXCL13 across cellular compartments and species. f, Dot plot presenting the scaled expression of CCL22, CXCL9 and CXCL13 across the cellular compartment of three different patients bearing HNSC tumors analyzed using scRNA-seq. For all box plot, boxes represent the 25th–75th percentile; the horizontal line represents the median; the whiskers represent 1.5 times the interquartile range (Tukey); and points represent individual samples. In c, statistical significance was calculated using a t-test with Bonferroni correction, *Padj ≤ 0.05, **Padj ≤ 0.01. All statistical tests were two-sided; P values are reported as exact values unless otherwise indicated.
Source data
In addition, the expression of CXCR3 and CXCR5 (two major drivers of T cell infiltration in tumors43,44,45,46) was conserved in murine T cells but the expression patterns of their canonical ligands were highly dissimilar across species. In murine TMEs, CXCL9 and CXCL10, and CXCL13, were enriched in the stroma (that is, fibroblasts), while biased toward myeloid and T/Treg cells, respectively, in human TMEs (Fig. 3d,e and Extended Data Fig. 3c,d). The T cell bias for CXCL13 expression in human TMEs was not absolute because occasional human tumors showed substantial stromal CXCL13 expression (Fig. 3f). These observations bear an immediate importance for the field as recent studies suggested that immune checkpoint responsive networks are organized around CXCL13+ T cells in patients47,48,49,50,51 and those rarely occur in mouse models. They also constitute important guidance for future mouse studies of human-relevant transcriptomic networks.
Interspecies deviations in TME immune cellular networks
Multiple studies have demonstrated that the presence of one cell type in the TME can correlate with another, typically because one either recruits or supports the other31,52,53. Therefore, we analyzed specific relationships in cell densities between cell types in human versus murine TMEs (Fig. 4). Total immune infiltration was inversely correlated with proliferating tumor cells (measured using Ki-67 staining; Extended Data Fig. 4a) in mice and human TMEs (Fig. 4a,b). Similarly, both species exhibited positive correlations between CD8 T cell frequencies and their degree of exhaustion (Fig. 4c and Extended Data Fig. 4b), and a relationship between cDC1 and cDC2 frequencies versus CD8 and CD4 T cell frequencies, indicative of co-maturation of these cells in tumors as described previously30,54 (Extended Data Fig. 4c).

a, Pearson correlation matrices (human on the left, hierarchically clustered; mouse on the right, ordered according to the human matrix), presenting correlations between the frequencies of different immune components in the TME. b–d, Side-by-side dot plots exemplifying the Pearson correlations shown in a of immune parameters between human (left, colored according to archetypes) and murine (right, colored according to tumor line) tumors. The specific correlations are indicated above each panel and grouped as conserved correlation on the left (b and c) and nonconserved correlation on the right (d). mac, macrophage. The solid lines indicate the linear fit (lm) and the shaded ribbons show the standard errors of the fitted lines (s.e. of the fit). Statistical significance was calculated using a Pearson correlation test with Benjamini–Hochberg correction, *Padj ≤ 0.05, **Padj ≤ 0.01, ***Padj ≤ 0.001, ****Padj ≤ 0.0001. All statistical tests were two-sided; P values are reported as exact values unless otherwise indicated.
Source data
However, several correlations differed between human and murine TMEs. For example, CD4 conventional T (Tconv) cell and Treg cell frequencies correlated in murine TMEs but not broadly in human TMEs (Extended Data Fig. 4d). More importantly, correlations between T cells and myeloid cells (Extended Data Fig. 4e) or between CD8 T cells and macrophages (Fig. 4d) in murine TMEs31 were not global features of human TMEs. Although this could partly originate from differential myeloid cell identification in human TMEs versus murine TMEs (human myeloid cells are sorted as HLA-DR+7 but not mouse myeloid cells), when restricting human samples to mouse-like immune-desert archetypes, the correlations were restored (insets in Fig. 4b–d and Extended Data Fig. 4c,e). This may indicate that these cells track one another but only under specific conditions, that is, in the absence of a large T cell pool or a global bias of the TME toward myeloid cells. Thus, these observations provide additional strong guardrails for interpreting murine tumor efficacy data for drug treatments that target these cell populations and their partners.
Consensus nonnegative matrix factorization identifies robust, cross-species transcriptomic programs
To compare human TMEs versus murine TMEs at a more granular and unbiased level, we applied a consensus nonnegative matrix factorization (cNMF) analytical pipeline5,55,56,57, anchoring on T cells and non-granulocytic myeloid cells to define their particular sets of GEPs and benchmark them cross-species (Fig. 5a).

a, Schematic of our analytical strategy using cNMF to compare the identity and coordination of GEPs in T cells and non-granulocytic myeloid cells across species. b,e, Heatmaps showing the Jaccard indexes used to quantify the similarity between human and mouse GEPs in T cells (b, using the top 20 genes per GEP) and myeloid cells (e, using the top 50 genes per GEP). Similarities of interest are highlighted in bold, colored fonts. c,d,f,g, Scatter plots showing the gene contribution (that is, gene weight) to human versus murine GEPs T_3 (c), T_9 (d), My_2 (f) and My_1 (g). Genes in bold show the highest overlapping contributions across species, while genes in gray have a higher contribution in one species versus the other. The dashed lines separate the 40 highest contributor genes from the others in either factor. h,i, Heatmaps showing the Jaccard indexes used to quantify the similarity between our human T cell (h) and myeloid cell (i) GEPs to the ones published in ref. 58. In b, e, h and i, statistical significance was calculated using a Fisher’s exact test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. All statistical tests were two-sided; P values are reported as exact values unless otherwise indicated.
Source data
When analyzing human T cells (Treg cells excluded7), we found nine stable GEPs (Extended Data Fig. 5a and Supplementary Table 2). We integrated Gene Ontology information with known genes functions to annotate these GEPs, such as human GEP T_3 was linked to ‘T cell cytotoxicity’ (PRF1, LAG3, GZMB and NKG7), while T_9 is linked to ‘CD4 regulation’ (TSC22D3, JUNB, RGS1, IL7R, CD69) (Extended Data Fig. 5c). The enrichment of some of these human GEPs correlated with TME composition (Extended Data Fig. 6a). For example, T_3 ‘T cell cytotoxicity’ and T_5 ‘CD4 T cell-associated’ correlated profoundly with CD8 and CD4 T cell frequencies, respectively. ‘CD4 regulation’ correlated with stroma and Treg enrichment in the tumor, possibly representing inhibitory circuits in TMEs. Repeating this analysis in murine TMEs uncovered 25 stable T cell GEPs (Extended Data Fig. 5e), similarly associated with T cell function and TME composition (Extended Data Figs. 5g and 6d). To quantify cross-species overlap of these GEPs, we used a Jaccard similarity index and found high degrees of similarity in three of the nine human T cell GEPs (Fig. 5b, Extended Data Fig. 6g,i and Supplementary Table 2). This notably included T_3 ‘T cell cytotoxicity’ (Fig. 5c) and T_9 ‘CD4 regulation’ (Fig. 5d).
Applying the same approach to the myeloid compartment in human TMEs identified 14 stable GEPs (Extended Data Fig. 5b). These included My_1 ‘inflammatory’ (IL1A, IL1B and NLRP3) and My_2 ‘IFN response’ (IFIT2, IFIT3, ISG15 and CXCL10) (Extended Data Fig. 5d and Supplementary Table 2). Some were highly correlated with cellular composition, including association of tumor-associated macrophage (TAM) densities with My_8 (‘lipid metabolism’) and monocyte densities with My_6 (‘migration’) and My_9 (‘regulation of defense response’) (Extended Data Fig. 6b). In murine TMEs, we identified 23 distinct and stable GEPs (Extended Data Fig. 5f), again often driven by genes known for their association with myeloid functions or whose enrichment correlated with TME composition (Extended Data Figs. 5h–j and 6e). Akin to T cells, a Jaccard analysis demonstrated high degrees of similarity for four pairs of myeloid GEPs (Fig. 5e and Extended Data Fig. 6h), including My_2 ‘IFN response’ (Fig. 5f) and My_1 ‘inflammatory’ (Fig. 5g), and the TAM-associated My_8 ‘lipid metabolism’ and My_11 ‘LYVE1 TAMs’ (Extended Data Fig. 6j,k).
To assess the robustness of the conserved T cells/myeloid GEPs, we used a Jaccard analysis with an independent dataset describing various GEPs (or ‘meta-programs’) across populations, studies and tumor indications in human58. All our cross-species conserved human GEPs (that is, My_1, My_2, My_8, My_11, T_3, T_4 and T_9) found an equivalent in this dataset (Fig. 5h,i). This was also the case when comparing these GEPs to another recent study describing T cells and myeloid GEPs in patients with glioma59 (Extended Data Fig. 7a), although sometimes our GEPs split into two in these other datasets, or conversely.
As the different platforms used to generate human and murine GEPs (bulk versus scRNA-seq, respectively) could cause the low degree of overlap seen in Fig. 5b,e, we used the recent ‘meta-programs’ study58 to compare scRNA-seq-generated GEPs in mouse versus human (Extended Data Fig. 7b). This both validated the conservation of GEPs described above and found additional new similarities, notably in T cells, by linking murine T_16, T_19, T_15 and T_23, respectively to ‘proliferative’, ‘naive’, ‘IFN-driven’ and ‘glycolytic’/’dysfunctional’ human T cell GEPs. In myeloid cells this also highlighted My_14/My_19 as similar to myeloid proliferative GEPs58. To this extent, GEP analysis may prove a substantially improved way to categorize cell functions both across studies and across species.
Leveraging the high resolution of our murine scRNA-seq datasets, we described the usage of murine GEPs by each murine cell subtype described in Extended Data Fig. 2. This underlined varying patterns of GEP usage across cell populations (Extended Data Fig. 7c), including some GEPs that are extremely specific to a given subtype (for example, Mu_T_25 in γδ T cells) while other GEPs seem to be used by many different cell subsets (for example, Mu_T_9 across all CD4 T cells).
We then asked whether the robust, conserved GEPs found across studies and platforms could also be observed in other biological processes than in tumors. Thus, we took advantage of a recent description of GEPs occurring over time and space in mouse myeloid cells during wound healing24 and again applied the Jaccard analysis approach (Extended Data Fig. 7d). This showed that three of these—My_1’inflammatory’, My_2’inflammatory’ and My_11 ‘LYVE1 TAMs’—had a similar GEP in the process of wound healing in mice.
This set of analyses overall defines a robust collection of core-conserved GEPs that recur from mouse to human across studies and biological processes. We hypothesize that these may serve as modern measures of immune identity, in some cases providing metrics for describing overall immune status and similarity, with cross-species conservation providing a tractable way to perturb and study the process in a model system.
Intercellular GEP ‘movements’ and their clinical relevance
We next examined coordinated correlations between T cell and myeloid GEPs24, which we called ‘movements’, as exemplar and because of the known importance of this axis for antitumor immune responses1,30,52,54. Several such ‘movements’ were present in human TMEs (Fig. 6a and Extended Data Fig. 6c–f), but only one was clearly conserved in murine TMEs: the association between T_3 ‘T cell cytotoxicity’ and My_2 ‘IFN response’. This conserved axis was particularly evident in IR CD8 mac and IS CD8 human archetypes and in MC38, CT26 and B16-F10 mouse models (Fig. 6b).

a, Heatmaps presenting the Pearson correlations of T cells versus myeloid GEP enrichment across human (left) or murine (right) TMEs. b, Scatter plots showing the correlation between the enrichments of GEPs T_3 and My_2 found across human (left, colored according to archetype) and mouse (right, colored according to tumor lines) tumors. Each dot represents a sample and the diagonal gray lines represent linear regressions. c, Scatterplot (corresponding to left of b) showing the binning of ImmunoProfiler patients as high or low for T_3 and My_2 GEPs, respectively (if present in the top or bottom 50% for each GEP enrichment). d, Kaplan–Meier graphs showing the overall survival of ImmunoProfiler patients stratified according to the binning shown in c. e, Scatterplot showing the relative enrichments of gene signatures calculated using the top 20 genes of GEPs T_3 and My_2 in patients from the TCGA. Patients were binned as high or low for each GEP if they were present in the top or bottom 50% for each calculated GEP gene score, respectively. Each dot represents a sample and the diagonal gray line represents the linear regression. f, Kaplan–Meier graph showing the overall survival of patients from the TCGA stratified according to the binning shown in e. In b, c and e, the solid lines indicate the linear fit (lm), the shaded ribbons show the s.e. of the fit. In a–c and e, statistical significance was calculated using a Pearson correlation test with Benjamini–Hochberg correction. *Padj ≤ 0.05, **Padj ≤ 0.01, ***Padj ≤ 0.001, ****Padj ≤ 0.0001. In d and f, statistical significance was calculated using a log-rank test. All statistical tests were two-sided; P values are reported as exact values unless otherwise indicated.
Source data
To assess the relevance of this GEP ‘movement’, we categorized patients as high or low for both T_3 ‘T cell cytotoxicity’ and My_2 ‘IFN response’ GEPs (Fig. 6c) and observed how their combinations parsed out overall survival. This showed a trend for improved survival of patients who displayed an overall high enrichment for both GEPs (T_3HiMy_2Hi) compared to any other combinations of these GEPs (Fig. 6d). Analyzing an independent transcriptomic dataset of whole human TMEs (TCGA7,41) demonstrated that these gene programs are also strongly correlated in this larger dataset (Fig. 6e). In this cohort, high T_3 ‘T cell cytotoxicity’ was generally associated with better outcomes compared to lower, but the T_3HiMy_2Hi condition is the most favorable for outcome compared to all the other combinations (Fig. 6f). As described elsewhere60,61, we found the relationship between cytotoxic T cells and IFN-stimulated myeloid cells to be beneficial for survival of patients with cancer.
Other human GEP correlations, such as T_9 ‘CD4 regulation’ with My_1 ‘inflammatory’, were not globally conserved in mice (Fig. 6a and Extended Data Fig. 7e,f). We again noted that when we censored human samples to include only those most similar to murine TMEs (ID CD4/CD8 mac), the correlation was less pronounced (Extended Data Fig. 7e). Interestingly, in the TCGA we found that patients with T_9Hi/My_1Lo gene expression levels tended to have improved survival compared to other conditions (Extended Data Fig. 7g,h). CD4 T cell activation in the absence of concurrent IL-1-related myeloid inflammation could thus be optimal, which is consistent with previous reports associating poor outcome and inflammatory myeloid cells62,63. We also noted that the mouse-like archetypes (generally T_9Lo) were among the worst surviving, hinting that mouse models may be relevant to study these classes of poor survivors in patients.
Altogether, these analyses showcase the use of GEPs and their correlations as ‘movements’ as entry points to studying cellular crosstalk in murine TMEs that is relevant to human TMEs and linked with patient outcomes.