
You have full access to this article via your institution.
Phosphatidylserine (PS) exposure on apoptotic cancer cells promotes their rapid and immunologically silent uptake by phagocytes, therefore favoring immune evasion. In a recent Nature study, evidence from viral infection models suggests that externalized PS also suppresses dendritic cell (DC) activation, adding to an expanding literature that identifies post-mitochondrial caspases as promising targets to inflame immunologically “cold” tumors.
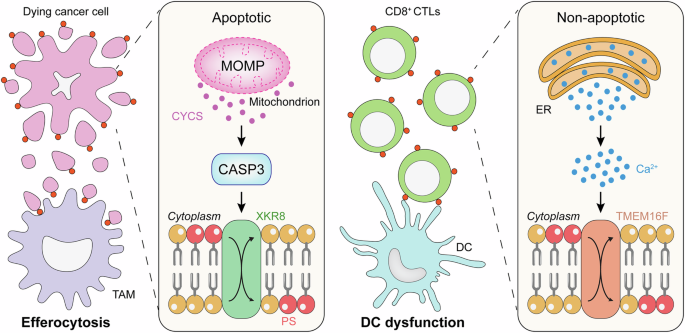
Mitochondrial apoptosis as dictated by mitochondrial outer membrane permeabilization (MOMP) and coordinated by caspases generally occurs in an immunologically silent manner.1 This reflects the ability of post-mitochondrial caspases, especially CASP3, to inactivate numerous proteins that would otherwise promote the emission of immunostimulatory signals by dying cells.1 Moreover, apoptosis-associated CASP3 activation favors the translocation of the phospholipid PS to the outer leaflet of the plasma membrane, which coordinates the rapid uptake of dying cells and their corpses by phagocytes while delivering powerful immunosuppressive signals.1 Accordingly, mice lacking a critical adaptor required for PS recognition by phagocytes exhibit an impaired uptake of cells undergoing apoptosis as part of physiological homeostasis and spontaneously develop autoimmune disorders.2
This is extraordinarily relevant for oncology at large, especially in the era of cancer immunotherapy. Malignant cells generally retain CASP3 competence in response to both natural or therapy-driven stress. However, when mitochondrial apoptosis is optimally engaged by widespread MOMP, CASP3 activation is dispensable for cell death, but critically regulates its kinetic and immunological consequences.1 Thus, blocking post-mitochondrial caspases and/or the immunosuppressive consequences of their activation represents a promising strategy to increase the immunogenicity of dying cancer cells. Further corroborating this notion, Medina and colleagues have recently harnessed models of chronic viral infection to demonstrate that PS exposure on the surface of CD8+ cytotoxic T lymphocytes (CTLs) promotes their exhaustion by impairing DC activity.3
Medina and colleagues investigated the functional significance of PS exposure on CD8+ T cells in a chronic murine LCMV infection model. Multiparametric flow cytometry revealed that antigen-specific CD8+ CTLs rapidly externalize PS upon activation, a process that becomes sustained under chronic antigenic stimulation. Integration of lipidomic and transcriptomic profiling indicated that this feature is embedded within a broad exhaustion-associated remodeling program.3 Consistently, high-dimensional flow cytometry and single-cell RNA sequencing demonstrated that PS-externalizing (PS+) CD8+ CTLs express co-inhibitory receptors like PD-1 and TIM-3 and exhibit transcriptional circuits linked to dysfunction. Moreover, PS+ CTLs displayed reduced proliferative capacity and impaired cytokine production upon restimulation, supporting the notion that PS exposure identifies a functionally compromised CD8+ CTL compartment.3
To determine whether externalized PS actively contributes to T cell dysfunction, Medina and co-authors employed an in vivo targeting strategy based on a PS-blocking monoclonal antibody. In chronically infected mice, this intervention promoted the expansion of virus-specific CD8+ T cells. Single-cell transcriptomics revealed that progenitor exhausted T cells, as identified by the co-expression of PD-1 and TCF-1, responded to PS blockage by undergoing a transcriptional reconfiguration indicative of enhanced proliferation, pointing to externalized PS as an inhibitor of self-renewal in CD8+ CTLs.3
Medina and colleagues next investigated whether externalized PS favors CD8+ CTL exhaustion via cell-intrinsic or extrinsic mechanisms. Data from CD8⁺ T cell and DC co-cultures and in vivo perturbation strategies demonstrated that PS exposure on CD8+ CTLs suppresses DC activity. More specifically, PS+CD8+ CTLs reduced the expression of co-stimulatory molecules and inflammatory cytokines by DCs, ultimately limiting their ability to sustain CD8⁺ CTL responses.3
To explore the therapeutic relevance of their findings, Medina and collaborators co-administered a PS-targeting antibody and an antibody specific for PD-L1 to LCMV-infected mice. This resulted in superior CD8+ CTL expansion and improved viral control over either treatment alone. Moreover, tumor-infiltrating PD-1+CD8+ T cells from cancer patients exhibited PS exposure, confirming the relevance of this mechanism for human disease and extending the breadth of these observations to oncology.3
Collectively, Medina and colleagues have unveiled a novel mechanism through which chronic viral infection promotes CD8+ CTL exhaustion, i.e., DC impairment as elicited by PS molecules externalized on the CTL surface.3 Importantly, PS exposure by CD8+ CTLs facing a chronic viral challenge could not be attributed to CASP3-dependent apoptosis, suggesting a role for activation-associated intracellular Ca2+ signaling and consequent phospholipid scrambling by TMEM16F.4 Notably, NK cells responding to malignant cells also appear to externalize PS along while acquiring a dysfunctional state.5 However, whether PS+ NK cells may also impair DC activity has not been investigated. Similarly, whereas Medina and colleagues pointed to a few surface receptors as potential culprits for DC dysfunction driven by PS+CD8+ CTLs, including molecules with an established DC-suppressing role like AXL and TIM4,6,7 functional experiments mechanistically establishing such connection have not yet been performed.
Intriguingly, defects in autophagy (a cytoprotective mechanism ensuring the degradation of unnecessary or potentially dangerous cytoplasmic entities) have been associated with deficient PS exposure on dying cells.8 These observations raise the intriguing possibility that blocking autophagy in malignant cells may not only enable superior anticancer immunosurveillance downstream of improved type I interferon signaling and MHC class I expression,9 but also as a consequence of limited PS exposure. This speculative hypothesis, however, has not yet been investigated in experimental tumor models.
Irrespective of these unknowns, as CD8+ CTL exhaustion is a key mechanism of cancer immune evasion,10 the findings from Medina and colleagues reinforce the notion that limiting the availability of surface-exposed PS in the tumor microenvironment represents a promising strategy to restore immunosurveillance (Fig. 1). That said, the PS-targeting antibody bavituximab failed to cooperate with docetaxel in patients with lung cancer enrolled in a Phase III clinical trial.11 Whether such a lack of efficacy originated from the ability of CASP3 to promote immunosuppression by PS-independent mechanisms, pointing to the blockade of post-mitochondrial caspases as to a superior immunostimulatory strategy for cancer therapy,1 remains unclear.

Cancer cells undergoing mitochondrial apoptosis expose phosphatidylserine (PS) on the cell surface, resulting in their rapid and immunologically silent disposal by tumor-associated macrophages (TAMs). Recent data from viral infection models suggest that externalized PS also promotes CD8+ cytotoxic T lymphocyte (CTL) exhaustion by inhibiting dendritic cells (DCs). Thus, blocking externalized PS or preventing its exposure by inhibiting CASP3, which has other immunosuppressive functions and is not required for cell death in the context of widespread mitochondrial outer membrane permeabilization (MOMP), represents a promising strategy to increase the immunogenicity of cancer cell death. Whether targeting TMEM16F, which underlies PS exposure in non-apoptotic settings, may have similar effects is unclear. ER, endoplasmic reticulum.