
Cell culture
Mouse melanoma cell line B16F10 (cat. SCSP-5233), mouse CRC cell line MC38 (cat. SCSP-5431), mouse HCC cell line Hepa1-6 (cat. SCSP-512), human CRC cell line SW620 (cat. TCHu101), human HCC cell line SNU-398 (cat. SCSP-5206), human melanoma cell line A375 (cat. TCHu155), and human embryonic kidney cell line 293T (cat. SCSP-502) were acquired from the National Collection of Authenticated Cell Cultures in Shanghai, China. B16F10, Hepa1-6, A375, and 293T cells were grown in Dulbecco’s modified Eagle’s medium (Gibco) containing 10% fetal bovine serum (FBS; Gibco). MC38 and SNU-398 cells were cultured in RPMI 1640 medium (Gibco) supplemented with 10% FBS. SW620 cells were cultivated in L-15 medium (Gibco) supplemented with 10% FBS. All cells were incubated at 37 °C in an environment containing 5% CO₂. Their authenticity was verified through short tandem repeat profiling and they were tested and determined to be free of mycoplasma contamination.
RNA extraction and quantitative PCR (qPCR)
Total RNA was extracted using TRIzol reagent (Invitrogen and Thermo Fisher Scientific). Subsequently, the isolated RNA was reverse-transcribed with HiScript III RT SuperMix for qPCR kit (cat. R323, Vazyme, Nanjing, China). qPCR was performed on a QuantStudio Real-Time PCR Instrument (Applied Biosystems and Thermo Fisher Scientific) using the ChamQ Universal SYBR qPCR Master Mix (cat. Q711, Vazyme). The primer sequences used in the experiment are presented in Supplementary Table 1. β-Actin was used as an endogenous control. Relative expression levels were determined by applying the 2−ΔΔCt method.
Expression vectors construction, short hairpin RNAs (shRNAs) synthesis, lentiviruses production, and stable cell lines constructions
To construct a vector expressing mouse SLC38A4, mouse Slc38a4 coding sequences (CDS) were amplified by PCR and subcloned into the BamHI and EcoRI sites of pcDNA3.1/V5-His B Vector (Invitrogen) using NovoRec® plus One step PCR Cloning Kit (Novoprotein, Suzhou, China). To generate mouse SLC38A4 expression lentivirus, mouse Slc38a4 CDS was subcloned into the NotI and NsiI sites of the lentiviral expression vector LV17 (EF-1a/Luciferase17&Puro) (GenePharma) using the ClonExpress Entry One Step Cloning Kit (Vazyme). The constructed LV17 vector was co-transfected with pGag/Pol, pRev, and pVSV-G into 293T cells using Lipofectamine 3000 (Invitrogen). Lentivirus was harvested 72 h after transfection and then filtered through 0.45 μm polyvinylidene fluoride filters. Human SLC38A4 expression lentivirus was generated, as described previously31. The CDS of mouse Myc and human MYC was PCR-amplified and subcloned into the EcoRI and XbaI sites of the pCMV-N-Flag Vector (Beyotime, Shanghai, China) using the NovoRec plus One step PCR Cloning Kit (Novoprotein) to construct mouse or human MYC expression vectors. The CDS of Cd24a was PCR-amplified and subcloned into the BamHI and EcoRI sites of pcDNA3.1/V5-His B Vector (Invitrogen) using NovoRec plus One step PCR Cloning Kit (Novoprotein, Suzhou, China). The primer sequences used for PCR are presented in Supplementary Table 1.
Two independent cDNA oligonucleotides suppressing mouse SLC38A4 expression were designed, synthesized, and subcloned into the lentiviral shRNA vector LV16 (U6/Luciferase17&Puro) (GenePharma, Shanghai, China), designated as mouse sh-SLC38A4-1 and sh-SLC38A4-2. cDNA oligonucleotides suppressing mouse CD24 expression were designed, synthesized, and subcloned into the lentiviral shRNA vector LV3 (H1/GFP&Puro) (GenePharma), designated as mouse sh-CD24. Scrambled non-targeting cDNA oligonucleotides were used as negative control (sh-NC). To generate mouse SLC38A4 or CD24 knockdown lentivirus, sh-SLC38A4-1, sh-SLC38A4-2, or sh-CD24 was co-transfected with pGag/Pol, pRev, and pVSV-G into 293T cells using Lipofectamine 3000 (Invitrogen). Lentivirus was harvested 72 h after transfection and then filtered through 0.45 μm polyvinylidene fluoride filters. The human SLC38A4 knockdown lentivirus was generated, as described previously31. Two independent cDNA oligonucleotides suppressing human or mouse MYC expression were designed, synthesized, and subcloned into the SuperSilencing shRNA expression vector pGPU6/Neo (GenePharma, Shanghai, China), designated as sh-MYC-1 and sh-MYC-2. The cDNA oligonucleotide sequences of mouse sh-SLC38A4-1, sh-SLC38A4-2, sh-CD24, human or mouse sh-MYC-1, sh-MYC-2, and sh-NC are listed in Supplementary Table 1.
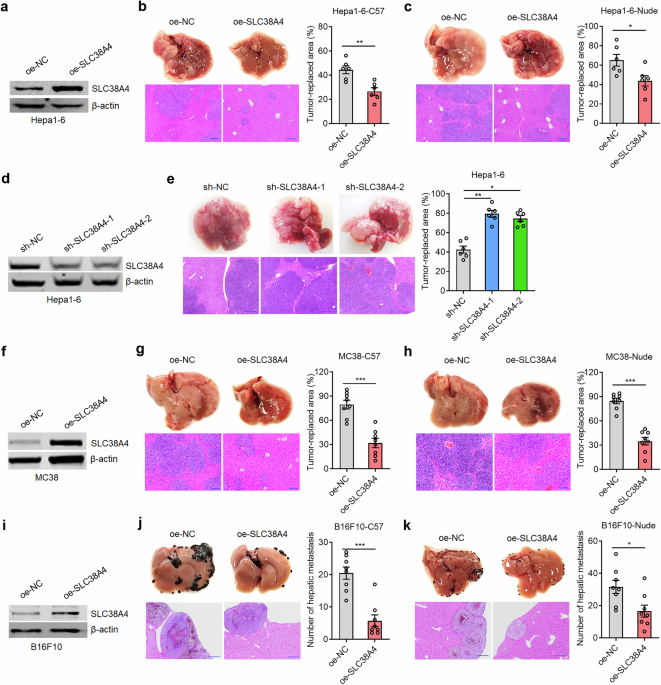
To generate cells stably overexpressing mouse SLC38A4, Hepa1-6, B16F10, and MC38 cells were infected with mouse SLC38A4 expression lentivirus in the presence of 8 μg/ml polybrene (Sigma-Aldrich). To establish cells stably overexpressing human SLC38A4, SW620 and SNU-398 cells were infected with human SLC38A4 expression lentivirus using 8 μg/ml polybrene (Sigma-Aldrich). To obtain cells with stable knockdown of mouse SLC38A4, Hepa1-6 cells were infected with mouse sh-SLC38A4-1, sh-SLC38A4-2, and sh-NC lentiviruses in the presence of 8 μg/ml polybrene (Sigma-Aldrich). Similarly, for the generation of cells with human SLC38A4 knockdown, SW620 cells and A375 cells were infected with human sh-SLC38A4-1, sh-SLC38A4-2, and sh-NC lentiviruses in the presence of 8 μg/ml polybrene (Sigma-Aldrich). Subsequently, these infected cells were selected with 2 μg/ml puromycin for 4 weeks.
To obtain cells with stable overexpression of mouse or human MYC, mouse or human MYC expression vectors were transfected into Hepa1-6 or SW620 cells, respectively, using Lipofectamine 3000 (Invitrogen), followed by being selected with 200 µg/ml neomycin for 4 weeks. To obtain cells with stable knockdown of mouse or human MYC, sh-MYCs were transfected into Hepa1-6, B16F10, MC38, SW620, SNU-398, and A375 cells using Lipofectamine 3000 followed by being selected with 200 µg/ml (for Hepa1-6, MC38, SW620, SNU-398, and A375) or 800 µg/ml (for B16F10) neomycin for 4 weeks.
To obtain cells with concurrent stable knockdown of SLC38A4 and CD24, Hepa1-6 cells with SLC38A4 knockdown were infected with sh-CD24 lentivirus in the presence of 8 μg/ml polybrene (Sigma-Aldrich), followed by being sorted by fluorescence-activated cell sorting (FACS) using BD FACS Aria II. To obtain cells with concurrent stable overexpression of SLC38A4 and MYC, MYC expression vectors were transfected into B16F10 and SW620 cells with SLC38A4 stable overexpression using Lipofectamine 3000 (Invitrogen), followed by being selected with 2 μg/ml puromycin and 800 µg/ml (for B16F10) or 200 µg/ml (for SW620) neomycin for 4 weeks. To obtain cells with concurrent stable knockdown of SLC38A4 and MYC, sh-MYCs were transfected into Hepa1-6 and A375 cells with SLC38A4 stable knockdown using Lipofectamine 3000, followed by being selected with 2 μg/ml puromycin and 200 µg/ml neomycin for 4 weeks.
Western blot
Total protein was harvested from the indicated cells using RIPA buffer (Beyotime) supplemented with a protease inhibitor cocktail (Calbiochem, Billerica, MA, USA). The cell lysates were then centrifuged at 12,000 rpm for 5 min at 4 °C. Equal amounts of protein were separated by SDS–PAGE and transferred onto nitrocellulose filter membranes. The membranes were blocked with 5% non-fat dry milk in PBS-T (Tween-20 at a concentration of 0.5 per thousand in PBS) at room temperature for 1 h. Then, they were incubated overnight at 4 °C in PBS-T with primary antibodies against mouse SLC38A4 (20857-1-AP, 1:500, Proteintech, Chicago, IN, USA), human SLC38A4 (ab58785, 1:1,000, Abcam, Cambridge, UK), or β-actin (66009-1-Ig, 1:10,000, Proteintech). After three washes, the membranes were further incubated with IRDye® 680RD goat antimouse IgG secondary antibody (926-68070, 1:10,000, Li-Cor Biosciences, Lincoln, NE, USA) or IRDye 800CW goat anti-rabbit IgG secondary antibody (926-32211, 1:10,000, Li-Cor). The protein bands were detected using an Odyssey infrared scanner (Li-Cor Biosciences). β-Actin was used as a loading control for total protein.
Animal studies
Male 6–8-week-old athymic BALB/c nude mice and C57BL/6 mice were obtained from the Shanghai Experimental Animal Center of the Chinese Academy of Sciences (Shanghai, China). All mice were maintained in a pathogen-free environment. To detect liver metastasis in vivo, indicated cells were intrasplenically injected into mice. At the specified time, the mice were sacrificed, and their livers were resected, fixed, and subjected to hematoxylin–eosin (HE) staining. The number or area of liver metastases was counted by researchers who were unaware of the experimental group allocation. Mouse experiments were carried out in accordance with animal welfare laws, guidelines, and policies and were approved by the Committee on Ethics of Medicine, Naval Medical University (Shanghai, China).
Kupffer cells isolation and in vitro phagocytosis analysis
Kupffer cells were isolated as described previously19. Briefly, liver tissues were perfused and then dissociated into single-cell suspensions. Hepatocytes were separated from single-cell suspensions by centrifugation at 40×g for 3 min. Subsequently, the supernatant was centrifuged at 650×g for 7 min to obtain non-parenchymal cells (NPCs). Finally, F4/80+ Kupffer cells were isolated from NPCs by magnetic-activated cell isolation (cat. 130-110-443, Miltenyi Biotech).
A total of 0.25 × 105 Kupffer cells co-cultured with 1 × 105 indicated tumor cells labeled with carboxyfluorescein succinimidyl ester (CFSE) using the CellTrace CFSE Cell Proliferation Kit (Thermo Fisher Scientific). The co-culture was carried out in ultra-low-attachment 96-well U-bottom plates (Corning) containing serum-free Iscove’s modified Dulbecco’s medium (Gibco). The cells were incubated at 37 °C in a humidified atmosphere containing 5% CO₂ for 2 h. Following co-culture, cells were collected and stained with an F4/80 antibody (cat. 17-4801-82, eBioscience). Flow cytometry was used to analyze the CFSE intensity within the F4/80+ cells. Phagocytosis efficiency was calculated as the ratio of Kupffer cells engulfed CFSE+ tumor cells. In CD24 neutralization experiments, 10 μg/ml blocking antibody against human CD24 (cat. 311101, BioLegend) or isotype control (mouse IgG2α, κ; cat. 400201, BioLegend) was added to SW620 cells 30 min before and throughout phagocytosis. Additionally, co-cultured Kupffer cells were pretreated with a mouse Fc-receptor blocking solution.
In vivo phagocytosis analysis
In vivo phagocytosis assays were performed as described previously19. Briefly, the indicated tumor cells, labeled with CFSE, were injected intrasplenically into the indicated mice. Twelve hours later, following perfusion, the livers of mice were dissociated into single-cell suspensions. Hepatocytes were removed from single-cell suspensions by centrifugation at 40×g for 3 min. NPCs were obtained by centrifugation of the supernatant at 650×g for 7 min. Flow cytometric staining was performed for viable NPCs (negative for 7-AAD; cat. 559925, BD Biosciences or negative for Zombie R718 Fixable Viability Dye; cat. 423115, BioLegend) to detect phagocytosis of CFSE-positive tumor cells by Kupffer cells (characterized as CD45+CD11bintF4/80+). This was achieved using antibodies against CD45.2 (cat. 109807, BioLegend), CD11b (cat. 101215, BioLegend), and F4/80 (cat. 123115, BioLegend).
Flow cytometry
Flow cytometry analysis was carried out using the Attune NxT Flow Cytometer in conjunction with Attune NxT Software version 5.2 (Thermo Fisher Scientific). The samples were detected using antibodies against human CD24 (cat. 311105, BioLegend), mouse CD24 (cat. 138503, BioLegend), mouse PD-L1 (cat. 124307, BioLegend), human PD-L1 (cat. 329705, BioLegend), mouse CD47 (cat. 127513, BioLegend), or human CD47 (cat. 323123, BioLegend). Data obtained from flow cytometry were processed and analyzed using FlowJo version 10.8.0. To ensure objectivity of the results, the investigators were blinded to the sample identities during flow cytometry.
Luciferase reporter assay
The promoter regions of mouse Cd24a (−800 bp to +100 bp) and human CD24 (−1,242 bp to −241 bp) were cloned into the XhoI and HindIII sites of the pGL3 basic vector (cat. 212936, Addgene), which is designed to express firefly luciferase. The resulting constructs were designated as pGL3-Cd24a promoter and pGL3-CD24 promoter, respectively. A mouse Cd24a promoter mutant was constructed by deleting the sequence from −321 bp to −312 bp, and a human CD24 promoter mutant was constructed by deleting the sequence from −1,173 bp to −1,164 bp. The primer sequences used in the experiment are presented in Supplementary Table 1.
The pGL3 vectors, SLC38A4 or MYC expression vector, and pRL-TK vector (Promega, Madison, WI, USA) expressing Renilla luciferase were co-transfected into 293T cells using Lipofectamine 3000 (Invitrogen). The pGL3 and pRL-TK vectors were co-transfected into the indicated tumor cells using Lipofectamine 3000 (Invitrogen). Forty-eight hours after transfection, luciferase activity was measured using the Dual-Luciferase® Reporter Assay System (Promega), following the manufacturer’s protocol. The relative firefly luciferase activity was normalized to Renilla luciferase activity.
Cleavage under targets and release using nuclease (CUT&RUN) assay
CUT&RUN assays were performed using the indicated cells. The assay used the Hyperactive pG-MNase CUT&RUN Assay Kit for PCR/qPCR (cat. HD101, Vazyme) and primary antibodies targeting MYC (cat. 13987, Cell Signaling Technology, Danvers, MA, USA). Enrichment of human CD24 and mouse Cd24a promoters was determined using qPCR. The primer sequences used are presented in Supplementary Table 1.
Tissue samples
Human HCC, primary CRC, and CRC liver metastatic tissues were collected from patients at Changhai Hospital and Eastern Hepatobiliary Surgery Hospital (Shanghai, China) with informed consent. This study was approved by the Committee on Ethics of Medicine, Naval Medical University (no. NMU-20231008).
Immunohistochemistry assay
Immunohistochemistry (IHC) staining was performed as described previously with a primary antibody against SLC38A4 (ab58785, 1:40, Abcam), CD24 (10600-1-AP, Proteintech, 1:600) for human clinical samples, CD24 (ab199140, 1:100, Abcam) for mice liver metastatic tissues, or MYC (ab32072, 1:50, Abcam)33. Slides were photographed using a Zeiss Axiophot photomicroscope (Carl Zeiss, Oberkochen, Germany).
Statistical analysis
All statistical analyses were performed using GraphPad Prism software (version 10.0). For comparisons, as specified in the figure legends, Student’s t test, one-way analysis of variance followed by Dunnett’s multiple comparisons test, Mann–Whitney test, Kruskal–Wallis test followed by Dunn’s multiple comparisons test, two-way analysis of variance followed by Tukey’s multiple comparisons test, and Spearman correlation analysis were conducted. Statistical significance was defined as P < 0.05.