
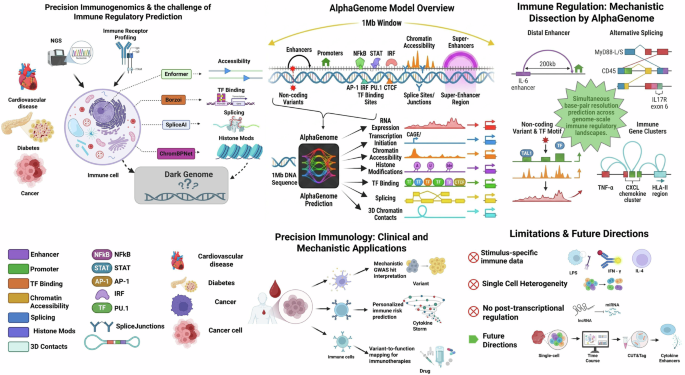
In a recent study published in Nature, Avsec et al.1 introduce AlphaGenome as a unified, multimodal framework that can predict thousands of functional genomic tracks and variant effects up to single-base-pair resolution from 1 Mb of DNA sequence.1 In the context of immune signaling, this is the first model capable of predicting immune regulation and immune-relevant splicing outcomes from DNA sequences alone by dissecting how non-coding variants shape receptor expression, transcription factor activation, enhancer-promoter communication, splicing of immune regulators, and chromatin accessibility in stimulus-responsive pathways.
The immune system plays a complex role in understanding the mechanistic development of several diseases, such as cancer, diabetes, and cardiovascular disorders. Immune signaling involves rapid, complex, and very often tissue-specific interactions. When it is combined with precision immunogenomics, it allows for studying how individual’s genes affect immune responses via high-throughput next-generation sequencing (NGS) and profiling tumor-specific mutations as well as immune cell receptors (TCR/BCR), thereby predicting highly tailored immunotherapies and treatment responses.2 Before AlphaGenome during the “Dark Genome” problem, studying immune signaling was extremely difficult. Immune pathways rely heavily on rapid transcriptional induction and alternative splicing driven by coordinated transcriptional factors (TFs) like nuclear NFκB, IRF, and AP-1 that do not operate in isolation but function as a coordinated network enabling immediate early responses, often transitioning into sustained inflammatory profiles.3 Upon stimulus-specific enhancer activation (LPS, DAMPs, and cytokines), there is alternative splicing of receptors and adaptors such as MyD88-L, MyD88-S, TLR adaptor isoforms, PTPN22 splicing variants, CD45 (PTPRC) isoforms (T cell activation), and IL17R exon 6 inclusion leading to chromatin remodeling at inflammatory loci and long-range enhancer-promoter loops such as IL-6, TNF-α, and CXCL chemokines. These processes are not governed by the coding regions, but by the non-coding cis-regulatory sequences.
Therefore, majority of immune phenotypes, including hyperinflammation, immunodeficiency, and autoimmunity, are driven by non-coding variants that modulate transcription factor binding motifs for NFκB, IRFs, STATs, and AP-1 chromatin accessibility at inflammatory enhancers, splicing of cytokines, receptors and adaptors, promoter usage and transcription initiation, and 3D chromatin architecture controlling cytokine gene clusters. All these modalities can be effectively predicted by AlphaGenome simultaneously at base-pair resolution, across 1 Mb of DNA sequence, which is precisely the scale at which immune regulatory elements operate. Non-coding variants are increasingly recognized as major contributors to immune-mediated diseases. Farh et al.4 demonstrated that over 90% of autoimmune-associated GWAS variants localize to enhancer regions in immune cells while Schmiedel et al.5 also reported that non-coding variants can modulate cytokine-responsive enhancer activity and transcription factor binding in T cells, directly shaping inter-individual immune responses highlighting the need for models capable of mechanistically interpreting non-coding immune regulatory variation.
The techniques employed before AlphaGenome relied on slower fragmented approach, often focusing on only one aspect of regulation at a time. NGS and RNA-Seq were used to measure RNA expression and identify variants. However, interpreting the functional consequences of non-coding variations was challenging. Traditional machine learning models such as Enformer and Borzoi lacked single-base resolution and could not process long DNA sequences efficiently while specialized tools including SpliceAI (splicing only) or ChromBPNet (chromatin accessibility only) offered no insight into how regulations are interconnected. For example, AlphaGenome exhibited a + 14.7% relative improvement in cell-type-specific gene-level-expression log-fold change prediction compared with Borzoi as well as specialized single-modality on their respective tasks, such as Orca on contact maps (contact map Pearson r + 6.3%; cell-type-specific differences +42.3%), ProCapNet on transcription initiation tracks (+15% total counts Pearson r), and ChromBPNet on accessibility (+1.6% for ATAC; +9.5% for DNase profile Jensen-Shannon divergence). It outperforms all existing models on eQTL direction prediction, splicing QTLs, chromatin accessibility QTLs, MPRA enhancer assays, and ClinVar non-coding variant interpretation. Moreover, time-consuming experiments like CRISPR gene-editing screens or reporter assays can confirm the effect of a mutation, but not for millions of potential variants.
AlphaGenome can predict multimodal RNA-Seq expression, CAGE and PRO-cap transcription initiation, DNase/ATAC accessibility, H3K27ac activation, histone modifications, TF binding (also immune TFs), splice sites, splice junctions, and 3D chromatin contact maps and loops at the same time, which directly map onto immune signaling biology and allow mechanistic interpretation of LPS-responsive and cytokine-inducible enhancers, variants altering inflammatory gene induction, and super-enhancers in macrophages and T cells. Immune genes are localized within dense regulatory neighborhoods. IL-6 has distal enhancer >200 kb away, while TNF-α locus spans ~100 kb of tightly interacting enhancers. Furthermore, CXCL chemokine clusters require long-range looping and HLA class II regulation spans hundreds of kilobases which are not captured by previous models. The 1 Mb input window of AlphaGenome allows it to model enhancer-promoter loops, super-enhancer architecture, chromatin domain boundaries, and multi-gene inflammatory clusters crucial for understanding immune signaling where distal enhancers determine the magnitude and kinetics of cytokine induction. While AlphaGenome is not specifically trained on immune cells, its TF binding prediction include NFκB (RELA, NFκB1, and NFκB2), IRF (IRF1, IRF3, and IRF3), STAT (STAT1, STAT3, and STAT5), AP-1 (cJUN and cFOS), PU.1 (SPI1), and CTCF (immune domain architecture) at 128 bp resolution ChIP-seq tracks derived from ENCODE across thousands of tracks where it effectively learns the immune regulatory code from more than 1600 human transcription factors including the immune-relevant TFs such as NFκB, IRFs, STATs, AP-1 through its TF ChIP-seq prediction heads which are the backbone of immune signal transduction (Fig. 1). For example, the model accurately predicts RELA occupancy at canonical kB enhancer motifs and reproduces STAT1 ChIP-seq binding profiles with high concordance to experimental data, outperforming prior multimodal models out of 1617 human TF ChIP-seq tracks reported. As shown in Fig. 6 of Avsec et al.1 paper, AlphaGenome captures the sequence determinants of immune-specific TF binding and can mechanistically interpret variants that alter NFκB enhancer or responsive activation. Furthermore, in the extended data figure, AlphaGenome achieves high Pearson correlation between predicted and observed STAT1 binding profiles by learning STAT-specific sequence determinants, which accurately outperforms Borzoi and Enformer models.

Schematic illustration of the unified multimodal prediction of immune regulation from 1 Mb DNA sequence by AlphaGenome. AlphaGenome provides an unprecedented framework for decoding immune regulatory logic by integrating long‑range chromatin interactions, transcription factor (TF) binding, splicing, and gene expression into a unified predictive model. Immune signaling pathways such as NFκB, STAT, IRF, AP-1, and PU.1 networks are governed by distal enhancers, promoter-enhancer looping, alternative splicing programs, and context‑specific chromatin accessibility. By modeling 1 Mb of sequence data, AlphaGenome captures distal enhancer-promoter communication essential for cytokine and chemokine gene regulations. For example, IL‑6 and TNF‑α enhancers often reside hundreds of kilobases from their promoters; AlphaGenome’s 3D contact predictions and TF-binding outputs allow these long‑range interactions to be inferred directly from sequence. Similarly, immune‑specific splicing events such as MyD88‑L/S isoform switching, IL7R exon 6 inclusion, and CD45 (PTPRC) alternative exons are predicted through integrated splice‑site, splice‑junction, and splice‑usage heads. These splicing programs are central to T‑cell activation, macrophage polarization, and cytokine signaling, and AlphaGenome’s multimodal outputs allow variant‑driven disruptions to be mechanistically traced. TF motif perturbations in immune enhancers, are exemplified by TAL1‑associated oncogenic variants that alter TF occupancy and chromatin accessibility. Because AlphaGenome predicts TF binding, accessibility, histone modifications, and transcription initiation jointly, it can distinguish whether a variant primarily affects motif strength, nucleosome positioning, enhancer activation, or downstream transcriptional output. This is particularly relevant for interpreting non‑coding variants in immune‑related diseases, where causal variants often reside in enhancer clusters such as the HLA‑II locus, the TNF‑α super‑enhancer, or the CXCL chemokine cluster. The translational applications of AlphaGenome include mechanistic interpretation of immune GWAS hits, variant-to-function mapping for immunotherapy response, and prediction of regulatory consequences in disease‑associated loci. Although AlphaGenome is not yet trained on stimulus‑specific immune datasets (e.g., LPS, IFN‑γ, IL‑4), its architecture provides a foundation for future models that incorporate dynamic immune activation states, single‑cell heterogeneity, and cytokine‑responsive enhancer landscapes (figure created in https://BioRender.com)
Avsec et al.1 discussed T-cell acute lymphocytic leukemia 1 (TAL1) comprehensively to illustrate a general principle highly relevant to immune signaling, although, TAL1 is a hematopoietic oncogene. A non-coding variant creates a de novo TF motif, which increases its TF binding and further enhances chromatin accessibility, enhancer activation, and its subsequent gene expression, which mimics how immune enhancers behave during toll-like receptor (TLR) activation, cytokine stimulation, T cell activation, and macrophage polarization. Interestingly, AlphaGenome can pinpoint enhancer variants that amplify cytokine storms, splicing variants that impair immune tolerance, promoter variants that reduce antiviral responses, and TF motif disruptions that alter macrophage polarization. By mapping variant-specific regulatory mechanisms, we can now understand JAK inhibitor responsiveness, anti-TNF therapy stratification, TLR4 pathway modulation, and splicing-modulation therapies which enables patient-specific immune regulatory profiling, mechanistic interpretation of GWAS hits, prediction of cytokine dysregulation risk, and mapping of immune cell-specific enhancer logic, a foundation of precision immunology (Fig. 1).
There are several immunology-specific limitations of AlphaGenome. It is not trained on stimulus-specific immune datasets such as LPS, IFN-γ, IL-4) nor does it incorporate single-cell immune heterogeneity. Moreover, it does not explicitly model cell-state transitions such as resting or activated states and also does not model post-transcriptional regulation such as miRNA and RNA-binding proteins. Furthermore, it does not include immune-specific epigenomic modalities such as CUT and RUN, CUT and Tag. However, it is a new model that should be able to incorporate macrophage activation time courses, immune-specific TF ChIP-seq datasets, single-cell chromatin accessibility, T cell differentiation trajectories, and cytokine-specific enhancer activation in the near future to create the first immune-specialized sequence-to-function model.