
Mice
Runx1+/WRPW embryos were generated by incorporation single-strand donor DNA into the Runx1 locus by the CRISPR/Cas9 genome editing technology. Zygotes generated from C57BL/6NJcl strain purchased from CLEA Japan were injected with mRNA encoding humanized S. pyogenes Cas9 that was invitro transcribed from pX330 plasmid (#42230, Addgene), single guide RNA (sgRNA) and single strand donor DNA, both of which were synthesized at Integrated DNA Technology. Similarly, Runx3WRPW, Runx3WRPF, Runx3WRPE mouse strains were generated by the CRISPR/Cas9 genome editing technology with appropriate mRNA for Cas9, sgRNA and donor DNA. Zygotes generated from C57BL/6NJcl for Runx3WRPW, Runx3WRPF and Runx3WRPE strains and ICR for Runx3WRPW strain purchased from CLEA Japan were injected with Cas9 mRNA, single guide RNA (sgRNA) and single-strand donor DNA. All these Runx3 mutant strains were established by crossing F0 founders with C57BL/6NJcl strain. In order to construct the target vector for R26lsl-R1, R26lsl-R1-WRPW, R26lsl-R1-WRPF, R26lsl-R3 and R26lsl-R3-WRPW mice, cDNA encoding WT and mutant Runx1 and Runx3 proteins were amplified by PCR to add AscI sites at the both ends, and these DNA fragments was ligated into an AscI-cleaved pCTV vector (#15912, Addgene). 30 μg of the target vector was linearized by AsiSI enzyme and transfected into the M1 ES cell line by electroporation as previously described5. After G418 selection, G418 resistant ES clones were screened for homologous recombination event by PCR. Appropriate ES clones were aggregated with blastocysts to generate chimera mice through which these mutant Rosa26 alleles were germline transmitted to the offspring, establishing the Rosa26 mouse lines. Runx1TY1 mice were generated by insertion of three copies of the TY1 (EVHTNQDPLD) tag followed by the hinge (GGG) region at the N-terminal end of P1-Runx1 protein by the CRISPR/Cas9 genome editing technology. Runx3Flag mice were generated by insertion of one copy of the Flag (DYKDDDDKLD) tag followed by the hinge (GG) region at the N-terminal end of P1-Runx3 protein by the CRISPR/Cas9 genome editing technology. R26DP-R3-WRPE allele was generated by CRISPR /Cas9 genome editing onto the R26lsl-R3 allele using in vitro fertilized zygotes between R26lsl-R3/lsl-R3 sperm and C57B6/N eggs. Runx1Y3F allele was also generated by CRISPR/Cas9-mediated genome editing. Sequences for sgRNA and donor DNA used in genome editing are listed in Extended Data Table 2. Cd4-Cre43, OT-I44, OT-II45, Thpok-Cre29, Zbtb7bΔS 7, Zbtb7bGFP 38 and TLE1/3/4TKO 9 mice have been previously described. Β2m-deficient mice (stock 002070) were from Jackson Laboratory. All mouse strains were bred and maintained in the animal facility at the RIKEN IMS. All animal procedures were in accordance with protocol AEY2022-019(2) approved by the institutional Animal Care and Use Committee of RIKEN Yokohama Branch. All the mice were euthanized with CO2 overdose following anesthesia using isoflurane. Data from both genders were combined for analysis, and unless otherwise specified, mice aged 4 to 14 weeks were analyzed.
Cells
Jurkat cells (RCB0806) and J.Cam1.6 cells were a gift from T. Saito at RIKEN IMS. Both Jurkat and J.Cam1.6 cells were maintained in RPMI-1640 medium supplemented with 10% FBS and antibiotics. pMYs retroviral vectors encoding TY1-tagged murine Runx1, Runx1WRPF or MigR1 retroviral vector encoding 3xFlag-TCF1 were transfected into GP2-293 cells (Clontech) with pVSV-G using FuGENE HD Transfection Reagent (Promega) according to the manufacturer’s protocol. Two days after transfection, supernatant was collected, passed through a 0.45 μm syringe filter and supplemented with 8 μg ml−1 polybrene (Sigma-Aldrich). Jurkat cells were suspended in the virus-containing medium at a density of 2.5 × 105 cells ml−1, and spin-infection was performed at 1,800 x g for 90 min at 32 °C.
Tissue processing
Thymus, spleen, lymph nodes and other organs were harvested from mice at 4 to 14 weeks of age and then mashed and passed through 100 μm pore cell strainer to make single-cell suspensions. After the hemolysis with ACK Lysing Buffer (Thermo Fisher Scientific), cells were washed with ice-cold staining buffer (D-PBS (-), 2 mM EDTA, 0.05% NaN3 and 2% FBS). Lamina propria lymphocytes from the small intestine were isolated as previously described23. Small intestine was cut into 5 mm pieces and incubated in 20 ml RPMI containing 2% FBS and 5 mM EDTA, with shaking at 200 rpm and 37 °C for 20 min. After incubation, tissue was washed with PBS twice by vortexing for 20 s to remove epithelial cells. The remainder of tissue was filtered and digested with RPMI with 2% FBS, 0.5 mg ml−1 collagenase IV (Sigma-Aldrich, C-5138) and 50 μg ml−1 DNase (043-26773; FUJIFILM) at 200 rpm and 37 °C for 30 min. Digested tissue was passed through a 100 µm strainer and subjected to Percoll gradient centrifugation at 1,800 rpm for 20 min using 40% and 80% Percoll in RPMI with 2% FBS. Cells located between the 40% and 80% Percoll layers were collected as LPLs.
Flow cytometry
Surface molecules were stained with specific antibodies by incubating for 30 min on ice. Following antibodies for surface molecules were purchased from BD Biosciences, BioLegend or Thermo Fisher Scientific; CD3ε (clone: 145-2C11), CD4 (clone: RM4-5), CD8α (clone: 53-6.7), CD19 (clone:6D5), CD24 (clone: M1/69), CD25 (clone: PC61.5), CD45 (clone: 30-F11), CD45.1 (clone: A20), CD45.2 (clone: 104), CD69 (clone: H1.2F3), CD117 (clone: 2B8), CD127 (clone: A7R34), CD135 (clone: A2F10), CD161 (clone: PK136), EpCAM (clone: G8.8), Ly-6G (clone: RB6-8C5), Integrin α4β7 (clone: DATK32), MHC-II (clone: M5/114.15.2), NKp46 (clone: 29A1.4), ScaI (clone: E13-161.7) TCRβ (clone: H57-597), and γδTCR (clone: GL3) and Vα2TCR (B20.1). In order to investigate intracellular molecules, cells were fixed and permeabilized by use of Transcription Factor Buffer Set (BD Biosciences) following surface staining. Following antibodies for intracellular molecules were purchased from BD Biosciences or Thermo Fisher Scientific; Zbtb7b (clone: 2POK), Runx3 (clone: R3-5G4), Rorγt (clone: B2D), PLZF (clone: 9E12) and Gata3 (clone: TWAJ). Dead cells were distinguished by use of 7-AAD (BD Biosciences) and LIVE/DEAD Fixable Dead Cell Stain (Thermo Fisher Scientific) for general and intracellular staining, respectively. Multi-color flow cytometric analysis was performed using BD FACSCanto II (BD Biosciences), and data was processed with FlowJo software (BD Biosciences). Cell subsets were sorted using a BD FACSAria III (BD Biosciences).
Cell cycle analyses
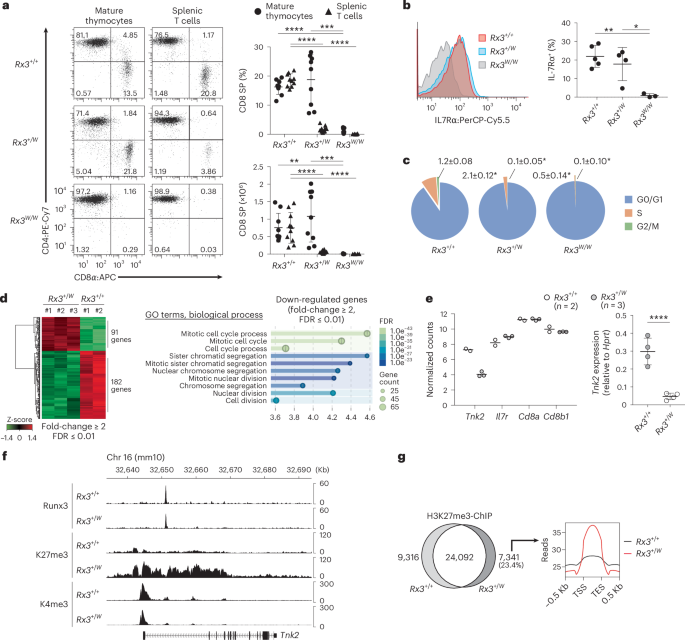
After the cell surface staining of freshly prepared whole thymocytes, cells were fixed with ice-cold 70% ethanol for 30 min on ice. After the meticulous cell wash using PBS, RNA was removed by the treatment of 0.1% Triton X-100 PBS containing 100 μg ml−1 Ribonuclease A (Thermo Fischer) for 30 min at room temperature. Intracellular DNA was stained using PBS (2% FCS) containing SYTOX Green (Thermo Fischer), and DNA content distribution was assessed by flow cytometry.
RNA sequencing
RNA was extracted from sorted CD8+ mature thymocytes of Wt and Rx+/W mice using the RNeasy Mini kit (Qiagen). Sequencing libraries were prepared using a NEBNext RNA Library Prep Kit for Illumina (NEB) according to the manufacturer’s protocol. Single-end 50 bp reads were obtained by Illumina HiSeq 2500. The reads were mapped to mm10 and analyzed with HISAT2 (v2.2.1) and edgeR (v3.36.0), respectively.
Bone marrow chimera
Bone marrow cells were collected from tibia and femur by flashing with RPMI. To generate bone marrow chimeras, recipient CD45.1+ ICR mice were irradiated at 950 rad. Donor cells were obtained from CD45.1+ ICR mice and CD45.1+/CD45.2+ F1 littermate mice, which were generated by crossing CD45.2+ C57BL/6 mice with CD45.1+ Rx3+/W mice in ICR background. The irradiated CD45.1+ ICR mice were reconstituted with a 50:50 mixture of CD45.1+ Rx3+/+ bone marrow cells and CD45.1+/CD45.2+ either Rx3+/+ or Rx3+/W bone marrow cells. Eight weeks after transplantation, chimeric mice were analyzed by flowcytometry to assess chimerism.
Cell sorting and enrichment
CD4 SP and CD8 SP thymocytes were enriched using EasySep Cell Separation with EasySep Mouse Streptavidin RapidSpheres Isolation Kit (STEMCELL Technologies). Thymi were harvested from C57BL/6NJcl mice at 3 to 5 weeks of age and mashed on a 100 μm pore cell strainer to obtain single-cell suspension. After the hemolysis with ACK Lysing Buffer (Thermo Fisher Scientific), cells were washed twice with D-PBS (-), and 108 cells were resuspended in 1 ml EasySep buffer (D-PBS (-), 2% FBS, 1 mM EDTA) at room temperature. The cell suspension was mixed with 50 μl normal rat serum and 10 μl biotin-conjugated anti-CD24 antibody (clone M1/69, BD Biosciences) with either 10 μl biotin-conjugated anti-CD4 (clone RM4-5, BioLegend) or CD8a (clone 53-6.7, BioLegend) antibody and incubated for 10 min at room temperature. The cell suspension was then mixed with 50 μl EasySep Mouse Streptavidin RapidSpheres and incubated for 5 min. After the reaction with RapidSpheres, the mixture was placed on the EasySep Magnet for 2.5 min, and the unlabeled cells were harvested. The purity the CD4 SP or CD8 SP thymocytes obtained was over 85%.
RNA isolation and quantitative PCR
DNaseI-treated total RNA was prepared from sorted CD8+ T cells using RNeasy mini kit (QIAGEN), and cDNA was synthesized by SuperScript IV reverse transcriptase (Thermo Fisher Scientific). Quantitative RT-PCR was performed using QuantStudio 3 Real-Time PCR system (Applied Biosystems) with Universal ProbeLibrary (Roche). The following primer sets and probes were used for Tnk2 and Hprt mRNA quantification: Tnk2-F, 5′-GGCCCTGCTCATCACAAA-3′, Tnk2-R, 5′-CCGTGATAGCTGTGCTCTGA-3′ and UPL #108. Hprt-F, 5′-TCCTCCTCAGACCGCTTTT-3′, Hprt-R, 5′-CCTGGTTCATCATCGCTAATC-3′ and UPL #95.
ChIP-seq and ChIP-qPCR
For ChIP-seq, MACS-enriched and FACS-sorted 5 × 106 Splenic CD8+ T cells were washed once with PBS supplemented with 2% FCS and cross-linked by incubation in a 1% formaldehyde solution for 10 min with gentle rotation at 22 to 26 °C. The reaction was quenched in 0.15 M glycine. Cells were then washed with ice-cold PBS containing 2% FCS for 10 min with gentle rotation at 4 °C and were lysed in Lysis Buffer 1 (50 mM HEPES pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% Glycerol, 0.5% NP-40, 0.25% Triton X-100) supplemented with cOmplete Protease Inhibitor Cocktail (Roche) for 10 min at 4 °C with gentle rotation. Nuclei were pelleted and were washed by Lysis Buffer 2 (10 mM Tris-HCl pH 8.0, 200 mM NaCl, 1 mM EDTA and 0.5 mM EGTA) supplemented with protease inhibitor cocktail. Pelleted nuclei were resuspended in 300 μl Lysis Buffer 3 (10 mM Tris-HCl pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% sodium deoxycholate and 0.5% N-laurylsarcosine sodium salt) supplemented with protease inhibitor cocktail and were sonicated 10 times using a model XL2000 ultrasonic cell disruptor (MICROSON) at output level 6 for 15 s. After removing debris by centrifugation, 30 μl 10% Triton X-100 was added to 270 μl supernatant, and sonicated chromatin was incubated overnight at 4 °C with anti-Histone H3K27me3 (clone C36B11, Cell Signaling Technology), anti-Histone H3K4me3 (clone C42D8, Cell Signaling Technology), or anti-RUNX3 (clone D6E2, Cell Signaling Technology) monoclonal antibodies, that were pre-conjugated with 50 μl Dynabeads M-280 Sheep anti-Rabbit IgG (Thermo Fisher Scientific). Magnetically collected beads were washed with ChIP-RIPA (50 mM HEPES (pH 7.6), 500 mM LiCl, 1 mM EDTA, 1% NP-40, 0.7% sodium deoxycholate) and TE buffer supplemented with 50 mM NaCl. Immunoprecipitates were eluted from beads into 100 μl of elution buffer (50 mM Tris-HCl pH 8.0, 10 mM EDTA, 1% SDS) by incubation for 15 min at 65 °C with vigorous shaking. Eluted immunoprecipitates were then incubated at 65 °C overnight for reverse-crosslinking. ChIP DNA samples were treated with RNase A (Thermo Fisher Scientific) at 37 °C for 1 h, followed by incubation with Proteinase K (Thermo Fisher Scientific) at 55 °C in the presence of 6 mM CaCl2 for 1 h and purified by phenol/chloroform extraction and ethanol precipitation. Purified DNA samples were subjected to re-sonication with a Covaris S220 to produce DNA fragments with an average size of 200 bp, and were used for library construction with NEBNext ChIP-seq Library Prep Master Mix set for Illumina Kit (NEB). Sequencing was performed by the RIKEN IMS sequence facility with Illumina HiSeq 1500. Sequencing data was analyzed on Galaxy platform (https://usegalaxy.org/). Adaptor-trimmed fastq files were mapped onto reference mouse genome (mm10) using ‘Bowtie2’ (v.2.5.3) with default setting. Generated bam datasets were processed with ‘Filter’ (v.2.5.2) and ‘MarkDuplicates’ (v.3.1.1.0) to remove reads with low mapping quality (<30), reads mapped on mitochondrial DNA, and PCR duplicates. Peaks were identified by use of ‘MACS2 callpeak’ (v.2.2.9.1). Bigwig files were generated from BedGraph using ‘Convert BedGraph to BigWig’ (v.1.0.1). ‘computeMatrix’ (v.3.5.4) and ‘plotHeatmap’ (v.3.5.4) were used to make heatmaps, and bigwig tracks were generated using ‘pyGenomeTraks’ (v.3.8).
For ChIP-qPCR, an equal number of CD4 SP and CD8 SP thymocytes were washed with D-PBS (-) twice, and the crosslink was performed by incubating in 50 mg ml−1 disuccinimidyl glutarate (Thermo Fisher Scientific) for 30 min at room temperature with gentle rotating followed by reaction with 1% paraformaldehyde (Sigma-Aldrich) for 10 min. The crosslink was quenched by adding 0.125 M glycine, and cells were rinsed with ice-cold D-PBS (-) containing 2% FBS. Cells were pelleted by centrifugation, and stored at −80 °C until the day of use. Pellet was defrosted on ice, suspended to Nuclear Isolation Buffer (50 mM Tris-HCl [pH 7.4], 60 mM KCl, 0.5% NP-40) supplemented with cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail (PI) (Sigma-Aldrich) and incubated for 10 min at 4 °C with gentle rotation to isolate nuclei. Nuclei were resuspended in 100 μl PI-supplied Lysis Buffer (50 mM Tris-HCl (pH 7.4), 10 mM EDTA, 0.5 mM EGTA and 0.5% SDS), and sonicated using Picoruptor 2 sonication device (Diagnode) with 16 cycles of output at high for 30 s followed by 30 s rest. Cell debris were removed by centrifugation for 10 min at maximum speed. Supernatant was transferred to clean tube, and 500 μl PI-supplied Dilution Buffer (20 mM Tris-HCl (pH 7.4), 150 mM NaCl, 2 mM EDTA and 1% Triton X-100) was added. The lysate was incubated with rabbit anti-TLE3 antibody (Proteintech, 11372-1-AP) or anti-Cbfb antibody pre-bound to Dynabeads Protein G beads (Thermo Fisher Scientific) overnight at 4 °C with gentle rotating. Chromatin-beads complex was washed with ChIP-RIPA Buffer (50 mM HEPES [pH 7.6], 500 mM LiCl, 1 mM EDTA, 1% NP-40, 0.7% Sodium Deoxycholate) > 5 times, and with TE buffer supplemented with 50 mM NaCl once. Chromatin-beads complex was resuspended to 100 ml of Elution Buffer (50 mM Tris-HCl [pH 8.0], 10 mM EDTA, 1% SDS) and incubated overnight at 65 °C with vigorous shaking. Samples were treated with RNaseA (Thermo Fisher Scientific) at 37 °C for 2 h followed by incubation with Proteinase K (Thermo Fisher Scientific) at 55 °C in the presence of 6 mM CaCl2 for 1 h. Precipitated chromatin DNA was purified using ChIP DNA Clean & Concentrator (ZYMO Research). Purified DNA was subjected to the quantification of E4, S4 region with PowerUp SYBR Green Master Mix (Thermo Fisher Scientific), QuantStudio 3 Real-Time PCR System (Thermo Fisher Scientific) and the following specific primers (Eurofins Genomics); E4p-F: 5′-TCTCCAAAGGGTAACAGGTGTCAG-3′, E4p-R: 5′-TGTGACTTACAAAGGTGCCTCCA-3′, S4-F: 5′-CCTTGTGTGGTCCCTCTCTTTG-3′, S4-R: 5′-GCAACAACCACCCTTCACAGG-3′, Alb-F: 5′-ACCTGCGTTACAGCATCCAC-3′, and Alb-R: 5′-TGCTGACAGAGCAGGAGACA-3′.
AlphaScreen
Recombinant proteins of mouse Runx1 and Runx3 were synthesized as N-terminal Flag-tagged and N-terminal single biotinylated form using a wheat cell-free protein synthesis system according to the procedures described previously25. To increase the yield and solubility of Runx proteins, self-annealed oligonucleotides encoding Runx binding sequence (sense, 5′-AGATGTGTGGTTAACCACAAAC-3′ and antisense, 5′-GTTTGTGGTTAACCACACATCT-3′) were added to the cell-free synthesis reaction46. In some reaction, the nucleotides in which mutations were subjected to one or both of the two Runx binding sequences were added. The sequences were as follows: M1: sense, 5′-AGATGTGTCCTTAACCACAAAC-3′ and antisense, 5′-GTTTGTGGTTAAGGACACATCT-3′, M2: sense, 5′-AGATGTGTGGTTAAGGACAAAC-3′ and antisense, 5′-GTTTGTCCTTAACCACACATCT-3′, and M3: sense, 5′-AGATGTGTCCTTAAGGACAAAC-3′ and antisense, 5′-GTTTGTCCTTAAGGACACATCT-3′ (where underline represents sequences that were mutated). These oligonucleotides were purchased from Thermos Fisher Scientific. A total of 497 human protein kinases containing 96 tyrosine kinases were synthesized as N-terminal FLAG-GST-tagged form using the same procedure used for Runx synthesis. In vitro binding assay using AlphaScreen was performed as previously described25 with slight modifications. Briefly, the binding reaction was performed in 15 µl binding buffer (50 mM Tris-HCl, pH 7.5, 1 mg ml−1 bovine serum albumin, 0.01% Tween20) containing 1 µl crude translation mixture of Flag-tagged and biotinylated proteins in a 384-well Opti-plate (PerkinElmer). The binding between two proteins was detected by anti-DYKDDDDK antibody (1E6, Wako) using AlphaScreen IgG (protein A) detection kit (Perkin Elmer). After 1 h incubation at room temperature, 10 µl detection mixture containing 7.5 ng anti-DYKDDDDK antibody (1E6, Wako), 0.08 μl streptavidin-coated donor beads and 0.08 μl protein A-conjugated acceptor beads (Perkin Elmer) were added to the binding rection in the Opti-plate. Binding between two proteins was detected with an EnVision multi-plate reader.
Mass spectrometry
Jurkat cells and J.CaM1.6 cells transduced with Ty1-tagged murine Runx1 or Runx1WRPF were treated with or without pervanadate (500 μM pervanadate, 3 mM hydrogen peroxide) for 15 min at 37 °C. Murine primary CD4 SP and CD8 SP thymocytes were prepared from Runx1Ty1/Ty1 mice expressing OT-II or OT-I transgenic TCR, respectively. The murine primary cells (107 cells) or the cultured cells (108 cells) were lysed with ice-cold RIPA buffer (20 mM HEPES-NaOH pH7.5, 1 mM EGTA, 1 mM MgCl2, 150 mM NaCl, 0.25% Na-deoxycholate, 0.05% SDS, 1% NP-40, Benzonase (Merck), PhosSTOP phosphatase inhibitor (Roche) and cOmplete protease inhibitor cocktail (Roche)). After the lysates were centrifuged at 20,000 × g for 15 min at 4 °C, the supernatants were incubated for 2 h at 4 °C with a 2.5 µl slurry of Sera-Mag SpeedBeads Protein A/G (Cytiva) pre-incubated with 1 µg anti-Ty1-tag mouse monoclonal IgG (Thermo Fisher Scientific, MA5-23513). The beads were washed four times with RIPA buffer and then twice with 50 mM ammonium bicarbonate. Proteins on the beads were digested by adding 200 ng trypsin/Lys-C mix (Promega) at 37 °C overnight. The resulting digests were reduced, alkylated, acidified, and desalted using GL-Tip SDB (GL Sciences). The eluates were evaporated and dissolved in 0.1% trifluoroacetic acid and 3% acetonitrile (ACN). LC-MS/MS analysis of the resultant peptides was performed on an EASY-nLC 1200 UHPLC connected to a Q Exactive Plus mass spectrometer through a nanoelectrospray ion source (Thermo Fisher Scientific). The peptides were separated on a C18 reversed-phase column (75 µm (inner diameter) × 150 mm; Nikkyo Technos) with a linear 4–32% ACN gradient for 0–100 min, followed by an increase to 80% ACN for 10 min and final hold at 80% ACN for 10 min. The mass spectrometer was operated in data-dependent acquisition mode with a top 10 MS/MS method. MS1 spectra were measured with a resolution of 70,000, an automatic gain control (AGC) target of 1e6 and a mass range of 350 to 1,500 m/z. HCD MS/MS spectra were acquired at a resolution of 17,500, an AGC target of 5e4, an isolation window of 2.0 m/z, a maximum injection time of 60 ms and a normalized collision energy of 27. Dynamic exclusion was set to 20 s. Raw data were directly analyzed against the SwissProt database restricted to Homo sapiens supplemented with Ty1-tagged murine Runx1 sequence using Proteome Discoverer v.2.4 (Thermo Fisher Scientific) with Sequest HT search engine. The search parameters were as follows: (i) trypsin as an enzyme with up to two missed cleavages, (ii) precursor mass tolerance of 10 ppm, (iii) fragment mass tolerance of 0.02 Da, (iv) carbamidomethylation of cysteine as a fixed modification and (v) acetylation of protein N terminus, oxidation of methionine and phosphorylation of serine, threonine and Y as variable modifications. Peptides were filtered at a false discovery rate of 1% using the Percolator node. Several selected peptides of Runx1, TLE3, LCK, FYN and ZAP70 were measured by parallel reaction monitoring (PRM)47, an MS/MS-based targeted quantification method using high-resolution MS. For quantitative analyses of phosphorylated versus unphosphorylated LEEVWRPY peptide of Runx1 protein, synthetic LEEAVWRPY and LEEAVWRPpY peptides (GL Biochem Japan) were measured to generate a standard curve. Targeted MS/MS scans were acquired by a time-scheduled inclusion list at a resolution of 70,000, an AGC target of 2e5, an isolation window of 2.0 m/z, a maximum injection time of 1 s and a normalized collision energy of 27. Time alignment and relative quantification of the transitions were performed using Skyline software.
Co-immunoprecipitation and immunoblotting
Peripheral CD8+ T cells were isolated form the spleens of Rx1Ty1/Ty1:Rx3Flag/Flag mice using EasySep Mouse CD8+ T Cell Isolation Kit (STEMCELL Technologies). Murine CD4 SP or CD 8SP thymocytes were obtained by beads cell sorting. Murine CD4 SP or CD 8SP thymocytes (106 cells), peripheral CD8+ T cells (107 cells) or Jurkat cells (107 cells) were lysed in RIPA buffer (20 mM HEPES-NaOH pH 7.5, 1 mM EGTA, 1 mM MgCl2, 150 mM NaCl, 0.25% sodium deoxycholate, 1% NP-40, Benzonase (Merck), PhosSTOP phosphatase inhibitor (Roche) and cOmplete protease inhibitor cocktail (Roche)). After centrifuging the lysates at 20,000 × g for 30 min at 4 °C, the supernatants were incubated for 4 h at 4 °C with 50 μl Protein A beads (Dynabeads) pre-incubated with 2.5 μg of anti-TLE3 antibody (Proteintech). The beads were washed four times with RIPA buffer, boiled in SDS-sample buffer and subjected to SDS-PAGE. Immunoblotting was performed using anti-Ty1 (Invitrogen, MA5-23513) to detect Ty1-Runx1, anti-Flag (Sigma-Aldrich: A8592) to detect 3xFlag-TCF1, and anti-Gapdh (Santa Cruz Biotechnology, sc-32233). Antibody binding was detected using ECL Prime Western Blotting Detection Reagent (Cytiva), and images were acquired on an Amersham Imager 680 (Cytiva). For AP treatment, 20 U alkaline phosphatase (Roche, 11097075001) was added to the Jurkat cell lysate and incubated at room temperature for 15 min.
Model building
To understand the dynamics and molecular interactions between TLE3 and the C-terminal WRPY-peptide of Runx1, the homology models of mouse TLE3 (mTLE3) and WRPY-peptide of mouse Runx1 (mRunx1) were first constructed using the crystal structure of human TLE-WD domain (hTLE) and the AlphaFold248 predicted model of human Runx1 (hRunx1), respectively. The crystal structure of the C-terminal SMWRPW peptide from the human Hes1 bound to the TLE-WD domain (PDB ID: 2CE9)11 was used as a template for the modeling of mRunx1 WRPY-peptide. For executing AlphaFold2, the run_docker.py python script was used to point to the FASTA file of the sequence, and max_template_date was set to a very recent date so that all the latest and up-to-date templates can be searched and used for model building. Structural superposition between mTLE3-hTLE1 and mRunx1-hRunx1 models was performed to evaluate the degree of similarity.
System preparation and all-atom MD simulations
The modeled mTLE3-mRunx1 WRPY-peptide complex structures were used for molecular dynamics (MD) simulations. To assess the impact of mutations, four mutated complexes were prepared and an additional system comprising only WRP-peptide of Runx1 was also prepared (Extended Data Table 1). The four mutated complexes harbored (i) WRPpY, (ii) WRPF, (iii) WRPE and (iv) WRPW peptides at the C terminus of Runx1 (where pY denotes phosphorylated tyrosine). For mutating the tyrosine residue and generating the mutant complexes (except WRPpY), the ‘swapaa’ tool of UCSF Chimera was used. For modeling the WRPpY structure, the UCSF Chimera package with SwissSidechain module was used to incorporate the nnAA-library, where the ‘swapnaa’ tool was used to select the chain ID, residue index and automatically assign the most favorable rotamer.
A total of six systems in addition to the WT complex (Extended Data Table 1) were used for MD simulations. The simulations of the complexes were carried out using GROMACS 5.1.2 software package with the CHARMM27 force field49. Each system was subjected to the addition of hydrogen atoms and subsequently solvated in a dodecahedron box of TIP3P water in the center at least 1.0 nm from the box boundary. The individual solvated system was then electrostatically neutralized by the addition of counterions, and subjected to 50,000 steps of energy minimization and an equilibration step consisting of a heating step from 0 to 300 K in 200 ps and a constant temperature phase at 300 K for 1 ns. The Parrinello-Rahman barostat pressure coupling was used to avoid the impact of velocity. Then, production runs were carried out for 200 ns for each system (total 1.2 µs) with periodic boundary conditions in an NPT ensemble with modified Berendsen temperature coupling and the constant pressure of 1 atm50. In this step, the LINCS algorithm was used to constrain the bond lengths, the Particle-mesh Ewald method was employed to calculate the electrostatic forces, the Fourier grid spacing and Coulomb radius were set at 0.16 and 1.4 nm respectively, and the van der Waals interactions were limited to 1.4 nm. The snapshots from the production MD were saved every 10 picoseconds for structural and dynamic analyses.
Analysis from MD simulations
The MD simulation trajectories of mTLE3-mRunx1 WRPY-peptide complex structures were analyzed using gmx rms and gmx gyrate GROMACS utilities to obtain the root mean square deviation and radius of gyration of each system, respectively. Various types of intermolecular interactions between mTLE3 and mRunx1 C-terminal peptides for all the molecular systems were computed using the Arpeggio web server51. The intermolecular hydrogen bond interactions formed between mTLE3 and mRunx1 C-terminal peptide for all the molecular systems were analyzed from the MD simulation trajectories using GetContacts (https://getcontacts.github.io/). The contacts were shown in a clustergram to make the interpretation clear for visualization.
Essential dynamics of mTLE3-mRunx1 C-terminal peptide complexes
The essential dynamics, which represent the principal motion directions by a set of eigenvectors, were performed on the MD trajectories of mTLE3-mRunx1 WRPY (WT), mTLE3-mRunx1 WRPpY, mTLE3-mRunx1 WRPF, mTLE3-mRunx1 WRPE, mTLE3-mRunx1 WRPW, and mTLE3-mRunx1 WRP complexes. In this analysis, a variance/covariance matrix was constructed by calculating the eigenvectors and eigenvalues, and their projection along the first two principal components was monitored by the PCA. The eigenvalues associated with each of the eigenvectors of mTLE3-mRunx1 WRPY (WT), mTLE3-mRunx1 WRPpY, mTLE3-mRunx1 WRPF, mTLE3-mRunx1 WRPE, mTLE3-mRunx1 WRPW, and mTLE3-mRunx1 WRP complexes were used to calculate the percentage of variability. The conformational changes associated with the FEL for the complexes were computed by gmx sham package52.
Binding free energy of mTLE3-mRunx1 C-terminal peptide complexes
The binding free energies between mTLE3 and mRunx1 C-terminal peptides for the mTLE3-mRunx1 WRPY (WT), mTLE3-mRunx1 WRPpY, mTLE3-mRunx1 WRPF, mTLE3-mRunx1 WRPE, mTLE3-mRunx1 WRPW, and mTLE3-mRunx1 WRP complexes were calculated using the molecular mechanics/Poisson Boltzmann surface area (MM/PBSA) methodology employed in the g_mmpbsa tool53 of GROMACS. The binding free energies of the complexes were computed as per the methodology described previously54. Finally, the binding free energy of complexes was calculated from 200 snapshots over the last 20 ns of the simulation trajectories as all the systems were stable during this time period.
In situ PLA
Total thymocytes and splenocytes were freshly isolated from mice, and specific populations were purified by sorting with BD FACSAria III as needed. Sample cells except derivates of cell lines and primary cells with GFP expression were stained with Alexa Fluor 488-conjugated anti-CD45 (Clone: 30-F11, Dilution: 1/200, BioLegend, 103122) antibody for 30 min on ice. Cells were washed with D-PBS (-) twice, resuspended to D-PBS (-) supplemented with 2% FBS at a density of 2 ×105 cells/ml, and 100 μl of the suspension was mounted on a glass slide by centrifugation with Cytospin 4 (Thermo Fisher Scientific) at 800 rpm for 5 min. Cells were fixed for 10 min with 4% paraformaldehyde in D-PBS (-) followed by the permeabilization for 10 min with 0.5% Triton-X-100. Glass slides were rinsed in D-PBS (-), and subjected to in situ PLA. After the blocking for an hour with supplied blocking buffer (Sigma-Aldrich), slides were incubated overnight with primary antibodies at 4 °C. Following antibodies were used; rabbit anti-TY1-tag polyclonal IgG (dilution: 1/100; GenScript, A01004), and mouse anti-FLAG-tag monoclonal IgG (Dilution: 1/100, Sigma-Aldrich, E3165), mouse anti-Lck monoclonal IgG (Dilution: 1/40, Santa Cruz Biotechnology, sc-433), rabbit anti-Bcl11b polyclonal IgG (dilution: 1/100; Bethyl Laboratories, A300-383A), rabbit anti-Cbfb polyclonal IgG (Dilution: 1/100, homemade), and mouse anti-Zap70 monoclonal IgG (dilution: 1/40; Santa Cruz Biotechnology, sc-32760). To generate mouse anti-VWRPY monoclonal IgG (dilution: 1/100, clone: Rp-6C2), 6xHis-tagged full-length human RUNX3 protein, expressed baculovirally in Sf9 cells and purified using a Ni-NTA resin (Superflow; QIAGEN), was used as an antigen to immunize mice55. Of the hybridoma clones producing pan-reactive antibodies to RUNX1, RUNX2 and RUNX3, the Rp-6C2 clones were found to be VWRPY specific, as revealed by western blotting36. Slides were washed with Wash Buffer A (Sigma-Aldrich) twice, and reacted with Duolink In Situ PLA Probe Anti-Mouse PLUS and Anti-Rabbit MINUS (Sigma-Aldrich) for 1 h at 37 °C followed by washing steps with Wash Buffer A. Ligation and amplification procedures were performed with Duolink In Situ Detection Reagents Orange (Sigma-Aldrich) according to manufacturer’s protocol. After the amplification step, samples were wash with 1x Wash Buffer B (Sigma-Aldrich) twice and once with 0.1x Wash Buffer B, and encapsulated with cover glass and Duolink In Situ Mounting Medium with DAPI (Sigma-Aldrich). Specimens were examined using BZ-X810 (Keyence) or ECLIPSE Ti-TIRF (Nikon) equipped with an sCMOS camera (ORCA flash 4.0, Hamamatsu Photonics), and densitometry was done with Image J software.
Data and statistical analyses
Data distribution was assumed to be normal, but this was not formally tested. Statistical analysis was performed by F-test and unpaired Students’ t-test with or without Welch’s correction using GraphPad Prism (v8.4.3) (GraphPad Software) or Excel (v2408) (Microsoft). The sequence information of the oligonucleotides is provided in Extended Data Table 2. The number of mice per group, the number of replicates per experiment, summary statistics and measures of dispersion are indicated in the legend of each figure. Mouse phenotyping was conducted under conditions that were largely randomized, and PLA experiments were fully randomized. Data collection and analysis were conducted without blinding to experimental groups because the mice had been genotyped before experimentation. No data were excluded from the analyses. Sample sizes were consistent with those commonly reported in the literature.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.