
Expression constructs
Expression constructs of indicated proteins were cloned into indicated vectors using PCR or the gateway system. Site-directed mutagenesis was performed by PCR to introduce desired amino acid substitutions. All expression constructs were sequenced by Seqlab.
Cell culture
HEK293T, U2OS, and HeLa cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin and maintained at 37 °C in a humidified atmosphere with 5% CO2. For Salmonella infections penicillin/streptomycin was omitted from the media. HeLa WT endogenous HA-GABARAPL2 cells were generated using CRISPR/Cas9 technology and kindly provided by the Behrends laboratory (LMU München, Germany). The guide RNAs used for the CRISPR/Cas 9 KO cells were designed using the GPP Web Portal of the Broad Institute (http://www.broadinstitute.org/rnai/public/analysis-tools/sgrna-design). These are: IRGQ #1: TTTGTGCTACCGGCGAACTG, IRGQ #2: GAATGCACTCAGTAAGGGAA, IRGQ #3: CGTGAGGCCTTTGAGACCGG, GABARAPL2 #1: GTCGAGCGAAATATCCCGACA, GABARAPL2 #2: GTCCCACAGAACACAGATGCG, GABARAPL2 #3: GGTTCCATCTGATATCACTG, and were cloned into Lentiviral vectors containing CAS9: pLenti-Puro or pLenti-Neo. This was done as described previously33: Lentiviral vectors containing Cas9 (pLenti-Puro, pLenti-Puro-EGFP, or pLenti-Neo) were digested with the restriction enzyme BsmBI and gel-purified. For ligation, 100 ng of the linearized vector was combined with 4 µl of annealed oligonucleotides (diluted 1:200) and incubated with T4 DNA ligase for 1 h at room temperature. The ligation products were subsequently transformed into XL1-Blue competent cells. Lentiviral particles were produced in HEK293T cells. Cells were seeded in six-well plates and cultured in 2 ml DMEM supplemented with 10% FBS until approximately 90% confluence. HEK293T cells were then co-transfected with three lentiviral plasmids encoding Cas9 and gene-specific sgRNAs (1.1 µg DNA per plasmid), together with the packaging plasmids pPAX2 (2.2 µg DNA) and pMD2.G (1 µg DNA). Viral supernatants were collected 24 h post-transfection, and the culture medium was replaced with 2 ml of fresh DMEM. A second collection was performed after an additional 24 h. The pooled viral supernatants (4 ml total) were centrifuged to remove cellular debris and stored at −80 °C. To generate knockout cell lines, target cells were infected with 1 ml of lentiviral supernatant containing three sgRNA constructs targeting the gene of interest. The remaining viral supernatant was stored at −80 °C. Forty-eight hours after infection, cells were selected in media containing 2 µg/ml of Puromycin or 1 mg/ml Neomycin. To create the reconstituted cell lines, HeLa GABARAPL2 KO cells (pLenti-Neo) were used and lentiviral transductions of HA-GABARAPL2 WT or mutants were performed, with a selection of 2 µg/ml of Puromycin or 1 mg/ml Neomycin.
CCCP was resuspended in DMSO and cells were treated with 40 μM for specific timepoints (30 min–4 h). Human IFNγ (AF-300-02; Peprotech) was added to cells for 24 h at a final concentration of 10 ng/ml. Nutrient starvation was achieved by replacing DMEM with EBSS (Gibco) for 30 min–4 h. BafilomycinA1 was resuspended in DMSO and cells were treated with 200 nM for specific timepoints. MRT67307 (a specific TBK1 inhibitor) was used at 5 μM for 4 h.
Plasmid transfections were performed with 3 μl GeneJuice (Merck Millipore), 0.5 μg plasmid DNA in 200 μl Opti-MEM (Life Technologies). After incubation for 15 min, the solution was added to the cells, which were lysed in lysis buffer or fixed with 4% paraformaldehyde 48 h later.
SiRNA transfections were performed with 3 μl RNAiMax (Invitrogen), 20 nM siRNA (IRGQ #1: TTTGTGCTACCGGCGAACTG, IRGQ #2: GAATGCACTCAG-TAAGGGAA, IRGQ #3: CGTGAGGCCTTTGAGACCGG; TBK1 #1: 5′-GACAGAAGUUGUGAUCACATT-3′) all purchased from Sigma) in 150 μl Opti-MEM (Life Technologies). After incubation for 30 min, the solution was added to the cells cultured in a 6-well dish, which were lysed in lysis buffer or fixed with 4% paraformaldehyde 72 h post transfection.
Salmonella infections
Salmonella SL1344 (WT) were streaked out on LB plates and single colonies were picked to inoculate 2 ml of LB media (containing appropriate antibiotics and 0.3 M NaCl) to grow at 37 °C for 16 h. The overnight culture was then diluted 1:33 in LB media (containing appropriate antibiotics and 0.3 M NaCl). After 2.5 h, the OD600 of Salmonella was determined and the cells were infected for 30 min with a MOI of 150 (considering that OD600 = 1 has ~1.3 × 109 bacteria/ml). The infection media was then exchanged to DMEM (+10% FBS) with 50 μg/ml Gentamycin and cells were lysed or fixed after indicated time points.
Immunofluorescence for confocal microscopy imaging
HeLa or U2OS cells were seeded onto glass coverslips in 12-well culture dishes and treated accordingly. Cells were washed in phosphate-buffered saline (PBS) before fixation with 4% paraformaldehyde for 15 min at room temperature. The coverslips were washed a further three times before permeabilization of the cells with 0.5% Triton X-100 in PBS for 10 min at room temperature. Cells were rinsed with PBS before being incubated for 1 h in 1% bovine serum albumin (BSA) in PBS for 1 h. Primary antibody incubation was done for 1 h in a humidified chamber with 1% BSA in PBS. After thorough washes in PBS, cells were incubated with secondary antibodies, 1% BSA in PBS for 1 h in the dark. Cells were washed three more times in PBS and once with deionized water before being mounted onto glass slides using ProLong Gold mounting reagent (Life Technologies), which contained the nuclear stain 4′,6-diamidino-2-phenylindole (DAPI). Slides were imaged using a Leica microscope Confocal SP 80 fitted with a 60x oil-immersion lens.
Immunofluorescence for Yokogawa CQ1 microscopy imaging
HeLa or T-REx mCherry-GFP GABARAPL2 WT, S10A and S10D mutant U2OS cells were seeded onto black, clear flat bottom 24- or 96-well plates (2000 cells/well for 96-well plates and 18,000 cells/well for 24-well plates). When indicated, cells were treated. Cells were washed in PBS before fixation with 4% paraformaldehyde for 15 min at room temperature. Cells were rinsed with PBS before being incubated for 1 h in permeabilization and primary antibody solution (0.1% Saponin (47036; Sigma), 5 mM MgCl2, 5% BSA in PBS). After washes in PBS, cells were incubated with Hoechst 33342 (R37605; Thermo Fisher), Alexa Fluor 647 Phalloidin (#8940; Cell Signaling Technology) and Alexa Fluor secondary antibodies in antibody solution (0.1% Saponin (47036; Sigma), 5 mM MgCl2, 5% BSA in PBS) for 1 h in the dark. Plates were imaged using a Yokogawa CQ1 microscope.
Cell lysis
For lysis, cells were washed with PBS and scraped on ice in IP lysis buffer (50 mM Hepes, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 25 mM NaF, 5% glycerol, 10 μM ZnCl2) or total cell lysis buffer (50 mM Tris HCl, pH 7.5, 1 mM EDTA, 1% SDS, 25 mM NaF, 1 μl/ml Benzonase (71205-25KUN; Millipore)), both supplemented with complete protease inhibitors (cOmplete, EDTA-free; Roche Diagnostics) and phosphatase inhibitors (P5726, P0044; Sigma). Extracts were cleared by centrifugation at 21000 × g for 15 min at 4 °C.
Immunoprecipitation of overexpressed proteins
Cleared cell extracts were mixed with HA-agarose beads (A2095; Sigma), Flag-M2 agarose beads (A2220; Sigma), RFP-Trap_A beads (rta-10; ChromoTek) or GFP-Trap_A beads (gta-10; ChromoTek) 16 h at 4 °C on a rotating platform. The beads were washed four times in IP lysis buffer. Immunoprecipitated and input samples were reduced in SDS sample buffer (50 mM Tris HCl, pH 6.8, 10% glycerol, 2% SDS, 0.02% bromophenol blue, 5% β-mercaptoethanol) and heated at 95 °C for 5 min34.
Immunoprecipitation of endogenous HA-GABARAPL2
HeLa cells stably expressing HA-tagged GABARAPL2 were seeded in 15-cm dishes and cultured to ~80% confluency prior to lysis. Cells were treated as indicated (e.g., siRNA knockdown, CCCP, or Salmonella infection). Cells were washed once with 10 mL PBS and lysed in 150 μL Total Cell Lysis (TCL) buffer (1% SDS, 50 mM Tris-HCl pH 7.5, 1 mM EDTA, 25 mM NaF, supplemented with protease inhibitor cocktails 2 and 3). Lysates were immediately transferred into 2 mL Eppendorf tubes, diluted with 1.35 mL of Normal Lysis (NL) buffer (150 mM NaCl, 50 mM HEPES pH 7.5, 1 mM EDTA, 1 mM EGTA, 10% (v/v) glycerol, 1% (v/v) Triton X-100, 10 μM ZnCl₂, 25 mM NaF, supplemented with protease inhibitor cocktails 2 and 3), and placed on ice. To degrade nucleic acids and reduce viscosity, 1 μL of Benzonase (Sigma–Aldrich) was added to each lysate, followed by incubation on ice for 20 min. Lysates were clarified by centrifugation at maximum speed for 10 minutes at 4 °C.
Input samples were prepared by transferring 50 μL of the cleared supernatant into a fresh tube, adding 10 μL of 4× SDS sample buffer, heating for 10 min at 95 °C, and storing at −20 °C.
HA immunoprecipitations were set up by incubating the remaining lysates with 20 μL of pre-equilibrated anti-HA magnetic beads (e.g., Thermo Fisher) overnight at 4 °C with gentle rotation in 1.5 mL tubes. The following day, beads were washed four times with chilled NL buffer. After the final wash, beads were dried using a 27 G ¾” needle to minimize residual buffer volume. Proteins were eluted by adding 20 μL of 2× SDS sample buffer directly to the beads, followed by heating at 95 °C for 10 min.
Protein binding assays
GST or GST-IRGQ were immobilized on glutathione-Sepharose beads (GE Healthcare) and combined with purified His-GABARAPL2 in protein binding buffer (150 mM NaCl, 50 mM, Tris, pH 7.5, 0.1% Nonidet P-40, supplemented with 5 mM DTT and 0.25 mg/mL BSA). The proteins were incubated on a rotating platform at 4 °C for 16 h. After five washes with buffer, proteins were diluted with SDS sample buffer (62.5 mM Tris-HCl pH 6.8, 10% (v/v) glycerol, 2% (w/v) SDS, 0.02% (w/v) bromophenol blue, 5% (v/v) β-mercaptoethanol), resolved by SDS-PAGE and analyzed by immunoblotting with the indicated antibodies.
Kinase assays
GABARAPL2 WT and mutant proteins were incubated in 20 μl phosphorylation buffer (50 mM Tris HCl, pH 7.5, 10 mM MgCl2, 0.1 mM EGTA, 20 mM ß-glycerophosphate, 1 mM DTT, 0.1 mM Na3VO4, γP32 ATP (500 cpm/pmol; SRP-201; Hartmann Analytic)) with 50 ng of recombinant GST-TBK1 for 15 minutes at 30 °C. The kinase assay was stopped by adding SDS sample buffer containing 1% β-mercaptoethanol and heating at 95 °C for 5 min. The samples were resolved by SDS-PAGE, and the gels were stained with InstantBlue (expedeon) and dried. The radioactivity was analyzed by autoradiography35.
Western blotting
For immunoblotting, proteins were resolved by SDS-PAGE and transferred to PVDF membranes. Blocking and primary antibody incubations were carried out in 5% BSA in TBS-T (150 mM NaCl, 20 mM Tris, pH 8.0, 0.1% Tween-20), secondary antibody incubations were carried out in 5% low-fat milk in TBS-T and washings in TBS-T. Blots were developed using Western Blotting Luminol Reagent (sc-2048; Santa Cruz). Immunoblot bands were quantified using ImageJ software. All Western blots shown are representative.
Antibodies
The following antibodies were used in this study: anti-HA-tag (11867423001; Roche), anti-FlagM2-tag (F3165; Sigma), anti-GFP-tag (Living Colors 632592; Clontech), anti-His-tag (11922416001; Roche), anti-vinculin (V4505; Sigma), anti-TBK1 (#3013; Cell Signaling Technology), anti-pTBK1 (pS172; #5483; Cell Signaling Technology), anti-IRGQ (HPA043254; Sigma), anti-GAPDH (#2118; Cell Signaling Technology), anti-GABARAPL2 (PM038; MBL), anti-LAMP1 (H4A3; DSHB), anti-p62 (M162-3; MBL), anti-IkBa (#9247; Cell Signaling Technology), anti-Histone H3 (ab1791, Abcam), anti-pSTAT1 (pY701; #7649; Cell Signaling Technology), anti-ATG3 (#3415; Cell Signaling Technology), anti-ATG7 (8558S, CST),anti-LC3B (PMO36; MBL), anti-ULK1 (8054S, CST), anti-MFN1 (14739S, CST), anti-pS10 GABARAPL2 (was generated by immunGlobe®, a chemically synthesized peptide (GABARAPL2 aa4-15) bearing a phosphate group at S10 (Ac-MFKEDH(pS)LEHRC-NH2) was used for immunization). Primary antibodies used for Western blotting were diluted 1:1000 and for immunofluorescence studies 1:200. Secondary HRP conjugated antibodies goat anti-mouse (sc-2031; Santa Cruz), goat anti-rabbit (sc-2030; Santa Cruz) and goat anti-rat (sc-2006; Santa Cruz) were used for immunoblotting. Anti-rat Alexa Fluor 647 (A-21247; Life Technologies), anti-rat Cy3 (712166153; Jackson Lab), anti-mouse Alexa Fluor 405 (A-31553; Life Technologies), anti-mouse Cy3 (715-165-151; Dianova), anti-mouse Alexa 647 (A-31626; Life Technologies) were used for immunofluorescence studies.
Mass spectrometry
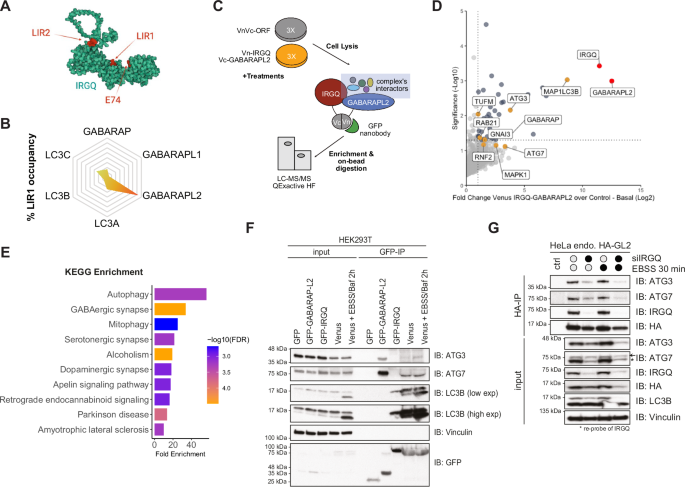
Cells were treated, lysed and IP was performed as stated above, after which trypsin digestion and peptide desalting were performed. The Venus IP dataset (Figs. 1C–E and S1F) had a total of 12 samples of which 3 were control IPs and 9 were 3 different conditions with 3 technical replicates each. Similarly, HA-IP dataset (Figs. 3C–E and S3D) had a total of 12 samples of which 3 were control IPs and 9 were 3 different conditions with 3 technical replicates each. Digested peptides were acidified with trifluoroacetic acid (TFA) (Sigma Aldrich) to inhibit trypsin and to acidify peptides for SDB-RPS StageTip desalting. Acidified peptides were loaded onto the SDB-RPS StageTips and then washed with 0.1% (v/v) TFA. Peptides were eluted using a two-step elution with 0.1% (v/v) TFA, 80% (v/v) ACN and then dried using a speed-vacuum concentrator (30–45 min at 45–60 °C). Dried peptides were stored at −20 °C.
Samples were analyzed on a Q Exactive HF coupled to an easy nLC 1200 (ThermoFisher Scientific) using a 35 cm long, 75 µm ID fused-silica column packed in house with 1.9 µm C18 particles (Reprosil pur, Dr. Maisch), and kept at 50 °C using an integrated column oven (Sonation). Peptides were eluted by a non-linear gradient from 4 to 28% acetonitrile over 45 min and directly sprayed into the mass-spectrometer equipped with a nanoFlex ion source (ThermoFisher Scientific). Full scan MS spectra (350–1650 m/z) were acquired in Profile mode at a resolution of 60,000 at m/z 200, a maximum injection time of 20 ms and an AGC target value of 3 × 106 charges. Up to 10 most intense peptides per full scan were isolated using a 1.4 Th window and fragmented using higher energy collisional dissociation (normalized collision energy of 27). MS/MS spectra were acquired in centroid mode with a resolution of 30,000, a maximum injection time of 110 ms and an AGC target value of 1 × 105. Single charged ions, ions with a charge state above 5 and ions with unassigned charge states were not considered for fragmentation and dynamic exclusion was set to 20 s.
MS raw data processing was performed with MaxQuant (v 1.6.5.0) and its in-build label-free quantification algorithm MaxLFQ applying default parameters. Acquired spectra were searched against the human reference proteome (Taxonomy ID 9606) downloaded from UniProt (21-11-2018; 94731 sequences including isoforms) and a collection of common contaminants (244 entries) using the Andromeda search engine integrated in MaxQuant. FDR was set to 1% on protein, PSM and site decoy level. Statistical analysis was done with Perseus 2.0.7.0. Proteins were defined as interactors, if they passed a 5% FDR corrected one sided two-sample T-test with a minimal enrichment factor of two. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (see Data Availability Statement).
Phos-tagTM SDS-PAGE
Phos-tagTM acrylamide (Wako) gels were used as indicated by the supplier. Gels were prepared with 10% acrylamide, 50 μM phos-tagTM and 100 μM MnCl2. Cells were lysed in SDS sample buffer supplemented with 10 μM MnCl2.
Proximity ligation assay (PLA)
HeLa cells were seeded onto black, clear flat bottom 96-well plates (2000 cells/well), treated with 10 ng/ml IFNγ for 24 h and infected with Salmonella. Cells were washed in PBS before fixation with 4% paraformaldehyde for 15 min at room temperature. Rabbit anti-IRGQ (HPA043254; Sigma) and mouse anti-HA (MMS-101P; Covance) were used with the respective Duolink in situ PLA probes (DUO92001, DUO92005; Sigma) and the PLA Duolink in situ detection reagent kit (DUO92008, Sigma) according to the manufacturer’s instructions. After washes in PBS, cells were incubated with Hoechst 33342 and Alexa Fluor 647 Phalloidin for 1 h in the dark. Plates were imaged using a Yokogawa CQ1 microscope and quantification of the PLA signal was performed using the Yokogawa CQ1 software.
Protein expression and purification
GST or His-tagged fusion proteins were expressed in E. coli strain BL21 (DE3). Bacteria were cultured in LB medium supplemented with 100 μg/mL ampicillin at 37 °C in a shaking incubator until OD600 ∼0.5–0.6. Protein expression was induced by the addition of 0.5 mM IPTG and cells were incubated at 16 °C for 16 h. Bacteria were harvested by centrifugation (3000 × g, 20 min) and lysed by sonication in GST lysis buffer (20 mM Tris HCl, pH 7.5, 10 mM EDTA, pH 8.0, 5 mM EGTA, 150 mM NaCl, 0.1% β-mercaptoethanol, 1 mM PMSF) or His lysis buffer (25 mM Tris HCl, pH 7.5, 200 mM NaCl, 0.1% β-mercaptoethanol, 1 mM PMSF, 1 mg/ml lysozyme). Lysates were cleared by centrifugation (28000 × g), 0.05% of Triton X-100 was added and the lysates were incubated with glutathione Sepharose 4B beads (GE Life Sciences) or Ni-NTA agarose beads (Thermo Fisher) on a rotating platform at 4 °C for 1 h. The beads were washed five times either in GST wash buffer (20 mM Tris HCl, pH 7.5, 10 mM EDTA, pH 8.0, 150 mM NaCl, 0.5% Triton X-100, 0.1% β-mercaptoethanol, 1 mM PMSF) or His wash buffer (25 mM Tris HCl, pH 7.5, 200 mM NaCl, 0.05% Triton X-100, 10 mM Imidazole). The immobilized proteins were reconstituted in GST storage buffer (20 mM Tris HCl, pH 7.5, 0.1% NaN3, 0.1% β-mercaptoethanol) or eluted with His elution buffer (25 mM Tris HCl, pH 7.5, 200 mM NaCl, 300 mM Imidazole) and dialyzed in (25 mM Tris HCl, pH 7.5, 200 mM NaCl) at 4 °C for 16 h. Recombinant GST-TBK1 was obtained from the MRC PPU DSTT in Dundee, UK (#DU12469)18.
FACS
T-REx mCherry-GFP GABARAPL2 WT, S10A and S10D mutant U2OS were cultivated in cell culture flasks until ~80% confluency and transferred to 6-well plates (500000 cells/well) for the experiments (3 replicates/condition for every cell line). Cells were treated overnight with doxycycline (final concentration: 1 µg/ml) to induce mCherry-GFP expression. Negative control was not induced. After induction, cells were washed once with PBS and treated as follows: untreated (stopped after 2 h), CCCP (40 µM, 2 h), EBSS (6 h) and EBSS + Bafilomycin (200 nM, 6 h). After treatments, cells were detached using trypsin/EDTA, centrifuged (5 min, 500 × g) and resuspended in 200 µl of FACS buffer (PBS + EDTA + FBS). FACS was performed directly at BD Symphony A5. Following singlet gating, cells were gated for high mCherry+ GFP+ cells using FlowJo software (version 10). The mean fluorescence intensity (MFI) for mCherry and GFP was calculated for each sample. Fold changes were calculated and normalized to the mean of each untreated cell line (WT, S10A, and S10D, respectively). Analysis was performed using GraphPad Prism software.
Modeling and simulations of IRGQ-ATG8 complexes
Full-length sequences of IRGQ (UniProtKB: Q8WZA9), GABARAPL2 (UniProtKB: P60520), MLP3B (LC3B, UniProtKB: Q9GZQ8), and ATG7 (UniProtKB: O95352) were used to model the 3D structures of various complexes using AlphaFold2-multimer model (AF2) multimers: (1) IRGQ-GABARAPL2, (2) IRGQ-GABARAPL2-ATG7, and (3) IRGQ-GABARAPL2-LC3B-ATG7. We obtained 100 models for each complex using the default AFv2.0 parameters (database updated, 2025). To evaluate the stability of the IRGQ-GABARAPL2 complex, we performed all-atom molecular dynamics (MD) simulations using the PDB structure of the IRGQ1-189-GABARAPL2 complex (PDB ID: 8Q6Q) as the initial model. Using the CHARMM-GUI server36, we refined the native (WT) IRGQ-GABARAPL2 complex and also modeled the phosphorylated version (S10PO4). We ensured that the disulfide bridge between C152 and C158 was preserved in our models. Protein complexes were placed in an octahedral box and solvated with the TIP3P water model and physiological salt concentration (150 mM NaCl). All-atom MD simulations of the IRGQ-GABARAPL2 complexes were performed with GROMACS (v 2021.5)37 using the CHARMM36m force field. Initially, the system was minimized using the steepest-descent algorithm until the maximum force reached 1000 kJ mol−1 nm−1. The equilibration phase was run in an NVT ensemble with the v-rescale thermostat at 310 K (τT = 1 ps)38. Position restraints were applied to the backbone (400 kJ mol−1 nm−2) and sidechain (40 kJ mol−1 nm−2) atoms to equilibrate the water. During the production run, the pressure was maintained at 1 bar (τP = 5 ps, compressibility = 4.5E-05 bar−1) using an isotropic c-rescale barostat39. Three replicates of production runs were simulated in each condition for 1000 ns with a 2 fs timestep. Coarse-grained MD simulations for the IRGQ-GABARAPL2 complexes were performed using (WT) and a phosphomimetic variant (S10D). The Martini force field was used to map the atomistic structure of the top-ranked AF2 model with the martinize2.py script40. For both variants, we employed the Go-Martini 3.0 model. Secondary structure assignments were done using DSSP, followed by automatic identification of disulfide bonds. Backbone restraints were applied with a force constant of 1000 kJ mol−1 nm−2, Go-like native contacts were modeled (ε = 12 kJ mol−1, residue distance cutoff = 3Å), along with predefined intrinsically disordered regions (regions 1–7, 179–191, 331–430, and 617–623) explicitly treated to preserve their conformational flexibility. The CG models were placed in a hexagonal simulation box, solvated with coarse-grained water beads, with 0.15 M NaCl. MD simulations were performed using GROMACS (v 2021.5)37. Systems were first energy minimized for 3000 steps with the steepest-descent approach, followed by an equilibration in an NPT ensemble at 310 K. Temperature and pressure were maintained, respectively, with a v-rescale thermostat (τT = 1 ps) and an isotropic c-rescale barostat (τP = 5 ps, compressibility = 4.5E-05 bar−1). During the production runs, we used the Parrinello-Rahman scheme (τP = 12 ps)41 to maintain system pressure. Simulations were performed with a 20 fs time step for 1000 ns, and 15 replicates were run for each system. Distances and contact maps for pairwise residue-residue interactions within each complex were computed using in-house scripts based on MDAnalysis v2.9.042. Contact maps were computed by counting pairwise residue contacts between chain A and chain B according to \({{{{\rm{AB}}}}}_{{{{\rm{cnts}}}}}=[{\sum }_{{{{\rm{i}}}}\in {{{\rm{A}}}}}{\sum }_{{{{\rm{j}}}}\in {{{\rm{B}}}}}{{{\rm{\sigma }}}}(|{{{{\rm{r}}}}}_{{{{\rm{ij}}}}}|)]\), where the sums extend over heavy atom positions of interacting residues \(({{{\rm{ij}}}})\; {{{\rm{and}}}}\,{{{\rm{\sigma }}}}(|{{{{\rm{r}}}}}_{{{{\rm{ij}}}}}|)=1- \left\lfloor 0.5-0.5(\tanh ((|{{{{\rm{r}}}}}_{{{{\rm{ij}}}}}|-{{{\rm{a}}}})/{{{\rm{b}}}}))\right\rfloor\)\(,\) a smooth sigmoidal counting function to limit interactions below the cut-off distance (\({r}_{{{{\rm{ij}}}}}\le a\,\)), where the cutoff parameter, \(a\) was set to 5 Å for atomistic simulations and 10 Å for CG simulations, while the smoothing parameter \(b\) was set to 0.5 and 1.0, respectively. Binding free energies for the WT and S10PO4 variant complex from atomistic simulations were estimated using the MMPBSA approach as implemented in the gmx_MMPBSA program with default parameters43. The binding free energy was decomposed into gas-phase and solvation contributions, with the gas-phase term (ΔGgas) comprising van der Waals and electrostatic energies, and the solvation term (ΔGsolv) including polar electrostatic contributions computed using the Poisson-Boltzmann model and nonpolar contributions estimated from solvent-accessible surface area. Per-residue free-energy decomposition was performed for both WT and S10PO4 systems to quantify residue-specific energetic contributions, and uncertainties were estimated from variability across independent replicas.
Statistical analysis
All experiments have a minimum of three biological replicates. Data are presented as the mean with error bars indicating the s.d. (standard deviation). Statistical significance of differences between experimental groups was assessed with Student’s t test. Differences in means were considered significant if p < 0.05. Differences with p < 0.05 are annotated as *, p < 0.01 are annotated as ** and p < 0.001 are annotated as ***. All western blots shown are representative of biological replicates. Immunofluorescent images were analyzed with CellProfiler 4.2.8.
Sequence alignments
Sequence alignments were performed using the Clustal algorithm44 with Ensemble identifiers. This approach allowed for the rapid and accurate alignment of protein sequences from the Ensemble database, facilitating the identification of conserved motifs and regions of interest.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.