
Construction and characterization of the bispecific cell engager MSLN×CD16A
To construct the bispecific cell engager MSLN×CD16A, we utilized the SpyTag/SpyCatcher protein conjugation system (Fig. 2a) to combine MSLN-SpyCatcher (for tumor localization and aggregation) and CD16A-SpyTag (for immune activation) at a molecular ratio of 1:1. Both MSLN-SpyCatcher and CD16A-SpyTag proteins were successfully induced and expressed in Escherichia coli Clear coli BL21 (DE3) and presented clear main bands by SDS‒PAGE (Supplementary Fig. S1a), with purities exceeding 90%, as determined by high-performance liquid chromatography (HPLC) (Supplementary Fig. S1b). Owing to potential inaccuracies in protein concentration measurements, the two proteins were mixed and coupled at different ratios, with a 1:1 molecular ratio used as the baseline. SDS‒PAGE analysis was performed to identify the optimal reaction conditions for obtaining the bispecific cell engager MSLN×CD16A (Fig. 2b, c). Flow cytometry analysis confirmed the binding of MSLN×CD16A to CD16A on CIML NK cells (Fig. 2d), with 10 µg of MSLN×CD16A reaching saturation binding to 1 × 10⁵ CIML NK cells (Supplementary Fig. S1c). Furthermore, both the flow cytometry and immunofluorescence staining results confirmed that MSLN×CD16A could specifically bind to MSLN-positive tumor cells (MKN45, NCI-N87, HGC27, and CFPAC-1) but did not bind to MSLN-negative tumor cells (MiaPaCa-2 and HuH-7) (Fig. 2e-i, Supplementary Fig. S1d). In silico analyses via the Immune Epitope Database (IEDB) suggest that the SpyTag/SpyCatcher system has low immunogenicity potential (Supplementary Fig. S1e, f).

Synthesis and binding characteristics of MSLN×CD16A. a Schematic diagram of MSLN×CD16A synthesis. Through the SpyTag/SpyCatcher protein conjugation system, MSLN×CD16A was synthesized with a molecular ratio of 1:1 between the target end MSLN-SpyCatcher and the effector end CD16A-SpyTag. b Visualization of the MSLN×CD16A structure via AlphaFold and PyMOL. c The target end MSLN-SpyCatcher and the effect end CD16A-SpyTag were mixed and coupled at different ratios with a 1:1 molecular ratio as the baseline to determine the optimal reaction conditions (left figure), with the red box indicating the optimal ratio. The right figure shows the SDS‒PAGE results for MSLN×CD16A. d Flow cytometry results demonstrating the binding of MSLN×CD16A to CIML NK cells. e Representative flow cytometry images of CD16A-SpyTag, MSLN-SpyCatcher, and MSLN×CD16A binding to MSLN-positive tumor cells (MKN45). f Mean fluorescence intensity of MKN45 in (e). g Binding of MSLN×CD16A to MSLN-positive (NCI-N87, HGC27, and CFPAC-1) and MSLN-negative (MiaPaCa-2, HuH-7) tumor cells. h Immunofluorescence staining to detect the binding of CD16A-SpyTag, MSLN-SpyCatcher, and MSLN×CD16A to MSLN-positive tumor cells (MKN45). CD16A-SpyTag, MSLN-SpyCatcher, and MSLN×CD16A were detected using His Tag antibody, red; cell nuclei, blue. i Representative confocal images showing the binding of MSLN×CD16A to MSLN-positive (NCI-N87, HGC27, and CFPAC-1) and MSLN-negative (HuH-7) tumor cells. For d and f, one-way ANOVA and Tukey’s multiple comparison test were used. Data represent mean ± SEM; n = 3 independent experiments. Scale bar: 50 µm. ns not significant; ∗P < 0.5; ∗∗P < 0.01; ∗∗∗P < 0.001; ∗∗∗∗P < 0.0001
Bispecific MSLN×CD16A specifically enhances CIML NK effector function in MSLN-positive tumor cells
To evaluate whether MSLN×CD16A induces CIML NK cell effector activation against MSLN-positive tumor cells in vitro, CIML NK cells were cocultured with various target cells for 6 h and treated with PBS or equal concentrations of the effector-end CD16A-SpyTag, target-end MSLN-SpyCatcher, or MSLN×CD16A. In the presence of MSLN-positive tumor cells (HGC27, MKN45, and NCI-N87), CIML NK cells cocultured with MSLN×CD16A presented significantly increased levels of degranulation, as assessed by CD107a expression (Fig. 3a, b). Additionally, MSLN×CD16A treatment significantly increased the production of the inflammatory cytokines IFNγ and TNFα by CIML NK cells (Fig. 3a, d, e). Furthermore, an increase in CD69, an activation marker for CIML NK cells, was observed in the MSLN×CD16A-treated group compared with the control group (Fig. 3a, c). However, this effect was not observed in CIML NK cells cocultured with MSLN-negative tumor cells (MiaPaCa-2 and HuH7) and treated with MSLN×CD16A (Fig. 3b-e).

MSLN×CD16A potently induces CIML NK cell activation and depletion of MSLN-positive target cells. a CIML NK cells were cocultured with MSLN-positive target cells HGC27 at an E:T ratio of 5:1 for 6 h and treated with PBS, CD16A-SpyTag, MSLN-SpyCatcher, or MSLN×CD16A to assess CIML NK cell expression of CD107a (a degranulation marker), CD69, intracellular IFNγ and TNFα. Representative flow cytometry images are shown in the upper panel. b–e Different MSLN-positive target cells (MKN45 and NCI-N87) and MSLN-negative target cells (MiaPaCa-2 and HuH7) were cocultured and treated as described in (a) to evaluate CIML NK cell functions, including CD107a, CD69, intracellular TNF-α and IFN-γ, via flow cytometry. f CIML NK cell cytotoxicity under different treatments was evaluated via a cell viability (green)/toxicity (red) assay after coincubation with HGC27-MCSs at an E:T ratio of 10:1. Representative confocal microscopy images of HGC27-MCS cells (upper panel) and corresponding surface display images (lower panel) are shown. g Quantitative analysis of live/dead cells in HGC27-MCS as shown in (f). h–l CIML NK cell cytotoxicity was measured under different treatment conditions at the indicated E:T ratios. MCS multicellular spheroids. For a–e and g, one-way ANOVA and Tukey’s multiple comparison test were used. For h–l, two-way ANOVA and Tukey’s multiple comparison test were used. The data are presented as the means ± SEMs; n = 3 independent experiments. Scale bar: 100 µm. ns not significant; ∗P < 0.5; ∗∗P < 0.01; ∗∗∗P < 0.001; ∗∗∗∗P < 0.0001
Most importantly, MSLN×CD16A mediated potent CIML NK cell cytotoxicity. In the multicellular spheroid (MCS) model established with the human gastric cancer cell line HGC27 (Fig. 3f), tumor cells in the control group remained mostly viable, whereas the proportion of surviving tumor cells in the MSLN×CD16A-treated group was significantly reduced, with a survival rate of 53% (Fig. 3g). Moreover, when CIML NK cells were cocultured with MSLN-positive tumor cells in vitro, MSLN×CD16A induced greater cytotoxicity, especially at high E:T ratios (Fig. 3h–j). However, MSLN×CD16A-mediated killing was specific, as it did not enhance CIML NK cell cytotoxicity against MSLN-negative cells (MiaPaCa-2 and HuH7) (Fig. 3k, l). These results demonstrate that MSLN×CD16A enhances the activation of CIML NK cells and imparts target-dependent cytotoxic activity to CIML NK cells.
Anti-MSLN CAR-like NK cells exhibit optimal activation and cytotoxic killing activity
When MSLN×CD16A is combined with CIML NK cell therapy to maximize effector activation and cytotoxic function, it is coincubated with CIML NK cells prior to infusion, allowing for the spontaneous anchoring of MSLN×CD16A on CIML NK cells, thereby constructing anti-MSLN CAR-like NK cells. To confirm the feasibility and functionality of anti-MSLN CAR-like NK cell therapy, CIML NK cells were incubated with varying concentrations of MSLN×CD16A for 1 h, followed by two washes with PBS before functional studies. Compared with MSLN×CD16A combined with CIML NK cell therapy (in vitro assay without washing after coincubation), anti-MSLN CAR-like NK cells demonstrated comparable efficiency in killing NCI-N87 (MSLN+) target cells, regardless of the MSLN×CD16A concentration (Fig. 4a). These results indicate that anti-MSLN CAR-like NK cells stably anchor MSLN×CD16A on their surface, maintaining consistent tumor cell recognition and killing capabilities.

Anti-MSLN CAR-like NK cells exhibit superior activity and stable tumor cell killing capacity. a MSLN×CD16A was coincubated with CIML NK cells for 1 h and then washed with PBS to construct anti-MSLN CAR-like NK cells, whereas those not washed were MSLN×CD16A combined with CIML NK cell therapy (NK + MSLN×CD16A). The cytotoxicity of the two treatments against NCI-N87 (MSLN+) target cells was compared at different MSLN×CD16A concentrations, with an E:T ratio of 10:1. b Changes in the fluorescence intensity of 100 μg/ml MSLN×CD16A on the surface of anti-MSLN CAR-like NK (1 × 106) and CIML NK (Ctrl NK) cells at different time points were evaluated by flow cytometry (left), along with representative flow cytometry images (right). c Comparison of the cytotoxicity for different treatments on HGC27-MCS at an E:T ratio of 10:1 over time. d Assessment of anti-MSLN CAR-like NK cell cytotoxicity at different time points after coincubation with NCI-N87 (MSLN+) at an E:T ratio of 5:1. e–h Expression of CD107a and CD69, as well as production of intracellular IFNγ and TNFα by CIML NK cells (Ctrl NK), non-targeted anti-non CAR-like NK cells, and anti-MSLN CAR-like NK cells, when cocultured with MSLN-positive target cells (CFPAC-1, NCI-N87, MKN45, and HGC27) and MSLN-negative target cells (MiaPaCa-2 and HuH7) at a 5:1 E:T ratio. The NK cells in the figures are all CIML NK cells. Ctrl NK: CIML NK cells; NK + MSLN×CD16A: MSLN×CD16A combined with CIML NK cell therapy. Two-way ANOVA and Tukey’s multiple comparison test were used for a, b and d. Data represent the mean ± SEM; n = 3 independent experiments. Scale bar: 100 µm. ns not significant; ∗P < 0.5; ∗∗P < 0.01; ∗∗∗P < 0.001; ∗∗∗∗P < 0.0001
To further evaluate the retention and activity of MSLN×CD16A on the surface of anti-MSLN CAR-like NK cells over time, 100 µg/ml MSLN×CD16A was coincubated with CIML NK cells for 1 h, washed, and cultured in medium for 1, 12, 24, or 48 h before detection. The flow cytometry results revealed that MSLN×CD16A remained on the surface of the anti-MSLN CAR-like NK cells for at least 48 h (Fig. 4b). Although the fluorescence intensity of MSLN×CD16A decreased after 48 h, likely due to partial internalization, it did not significantly affect the cytotoxic activity of the anti-MSLN CAR-like NK cells. In the HGC27-MCS model, the cytotoxic ability (approximately 50%) of the washed anti-MSLN CAR-like NK cells remained stable over time (Fig. 4c, Supplementary Fig. S1g). Similarly, the flow cytometry results revealed that, compared with CIML NK cells, anti-MSLN CAR-like NK cells exhibited significantly greater cytotoxicity against NCI-N87 (MSLN+) targets at all time points, with no significant difference from MSLN×CD16A combined with CIML NK cell therapy (Fig. 4d). These findings confirm that anti-MSLN CAR-like NK cells retain their ability to kill tumor targets over time, demonstrating the feasibility of this type of cell therapy.
Next, we evaluated the specific antitumor activity of anti-MSLN CAR-like NK cells. After 6 h of coculture, compared with CIML NK cells and anti-non CAR-like NK cells, anti-MSLN CAR-like NK cells presented significantly increased expression of CD107a and CD69, and increased IFNγ and TNFα production when cocultured with MSLN-positive target cells, including CFPAC-1, NCI-N87, MKN45, and HGC27 cells (Fig. 4e-h). In contrast, anti-MSLN CAR-like NK cells were not significantly activated by MSLN-negative cells (MiaPaCa-2 and HuH7).
CAR-like NK cell therapy targeting MSLN demonstrates optimal tumor growth inhibition in solid tumors
In vitro experiments indicate that CAR-like NK cell therapy is feasible. After washing, anti-MSLN CAR-like NK cells recognize and kill tumor cells due to the stable anchoring of MSLN×CD16A on their surface, with an efficacy comparable to that of MSLN×CD16A combined with CIML NK cell therapy. However, the antitumor efficacy of anti-MSLN CAR-like NK cells in vivo remains unclear. Therefore, we further evaluated its antitumor effect in vivo by establishing an MSLN-positive MKN45 xenograft mouse model (Fig. 5a). When the tumor volume reached 100 mm3, the mice were treated twice with PBS, 100 µg MSLN×CD16A, 1 × 107 CIML NK cells (Ctrl NK), MSLN×CD16A combined with CIML NK cells (NK + MSLN×CD16A), or anti-MSLN CAR-like NK cells (at 7-day intervals). As shown in Fig. 5b, c, in the anti-MSLN CAR-like NK group, the tumor volume remained relatively stable at a relatively low level, whereas the tumor burden in the PBS and MSLN×CD16A groups increased dramatically. Although CIML NK cell therapy delayed tumor growth, no significant difference was observed compared to the PBS control group. Notably, anti-MSLN CAR-like NK cells demonstrated a more significant advantage in inhibiting tumor growth than MSLN×CD16A combined with CIML NK cell therapy did (P < 0.05), with this trend strengthening over time. These findings suggest that MSLN×CD16A combined with CIML NK cell treatment (CAR-like NK cells assembled in vivo) is not as effective as anti-MSLN CAR-like NK cells assembled in vitro in terms of antitumor effects.

Anti-MSLN CAR-like NK cells exhibit tumor inhibition and prevent tumor cell dissemination in a preclinical xenograft mouse model. a Schematic diagram of the treatment regimen for MKN45 tumor-bearing BALB/c nude mice. The mice were subcutaneously implanted with 6 × 106 MKN45 cells. When the tumor volume reached approximately 100 mm3, the mice were randomly grouped. PBS (control), MSLN×CD16A (100 µg), CIML NK (Ctrl NK, 1 × 107), MSLN×CD16A combined with CIML NK cell therapy (NK + MSLN×CD16A), and anti-MSLN CAR-like NK (1 × 107) were administered via tail vein injection. Recombinant human IL-2 (50,000 U) was administered intraperitoneally every other day to support the transferred NK cells. Figures were created with BioRender.com and SciDraw. b Tumor images harvested from different groups at the study endpoint. c Tumor kinetics in MKN45 tumor-bearing mice following treatment over time (n = 6). d Tumor volume curves of mice subcutaneously injected with NCI-N87 cells (n = 6). e Schematic diagram of the treatment regimen for the gastric cancer lung metastasis xenograft model. The lung metastasis model was established by injecting 3 × 106 MKN45 cells into BALB/c nude mice via the tail vein. On days 0 and 7, the tumor-bearing mice were treated with PBS (control), unanchored CIML NK cells, untargeted anti-non CAR-like NK cells, or anti-MSLN CAR-like NK cells (1 × 10⁷) via intravenous reinfusion, with 50,000 U of recombinant human IL-2 every other day. The tumor burden was assessed via BLI on days 0, 7, and 14. Figures were created with BioRender.com and SciDraw. f Tumor burden monitored at designated time points via BLI after the corresponding treatments (n = 5). g Quantification of tumor bioluminescence images at different time points in (f). h Schematic diagram of the treatment regimen for MSLN-high- and low-expressing peritoneal disseminated tumors. The mice were intraperitoneally injected with 5 × 10⁶ MSLN-high-expressing tumor cells (MKN45, NCI-N87) or MSLN-low-expressing tumor cells (MiaPaCa-2), followed by an intraperitoneal injection of 1 × 10⁷ CIML NK cells (Ctrl NK group) or anti-MSLN CAR-like NK cells, with recombinant human IL-2 given every other day. The tumor burden was monitored at designated time points via BLI. Figures were created with BioRender.com and SciDraw. i–k Quantification of the bioluminescence levels of MSLN-high- and MSLN-low-expressing peritoneal disseminated tumors at different time points in (l–n). l–n Tumor burden was monitored via BLI in peritoneal disseminated tumor-bearing mice treated with the corresponding cells. BLI: bioluminescence imaging. Two-way ANOVA and Tukey’s multiple comparison test were used. The data represent the means ± SEMs. ns, not significant; ∗P < 0.5; ∗∗P < 0.01; ∗∗∗P < 0.001; ∗∗∗∗P < 0.0001
To further validate the enhanced function of anti-MSLN CAR-like NK cells in a systemic environment, we conducted experiments using the NCI-N87 gastric cancer subcutaneous tumor model (Supplementary Fig. S2a). Compared with the PBS control, both types of NK cell therapy (NK + MSLN×CD16A and anti-MSLN CAR-like NK) significantly delayed tumor growth (Fig. 5d and Supplementary Fig. S2c). The average tumor weight in the anti-MSLN CAR-like NK group was 0.29 g, which was significantly lower than that in the PBS group (1.07 g) and the NK + MSLN×CD16A group (0.64 g) (Supplementary Fig. S2d). However, anti-MSLN CAR-like NK cells demonstrated a greater advantage in reducing the tumor burden (Supplementary Fig. S2e). In summary, anti-MSLN CAR-like NK cells not only demonstrate therapeutic feasibility but also exhibit unique antitumor advantages, providing new insights for tumor immunotherapy.
To assess the safety of anti-MSLN CAR-like NK cells, H&E staining of major organs, including the heart, liver, spleen, lung, and kidney, was performed on MKN45 tumor-bearing mice in each group (Supplementary Fig. S3a). No significant tissue damage or structural changes were observed. Additionally, routine blood tests and serum biochemical analysis indicated no obvious toxicity following anti-MSLN CAR-like NK cell therapy (Supplementary Fig. S3b). Furthermore, no significant body weight fluctuations were observed in the MKN45-bearing mice (Supplementary Fig. S3c) or NCI-N87-bearing mice (Supplementary Fig. S2b). These results show that anti-MSLN CAR-like NK cell therapy not only possesses excellent tumor-killing capacity but also demonstrates superior safety, offering broad potential for clinical application.
Anti-MSLN CAR-like NK cells exhibit robust antitumor efficacy and specificity in intraperitoneal and pulmonary metastatic tumor models
To evaluate whether anti-MSLN CAR-like NK cells can inhibit distant tumor metastasis in gastric cancer, we established a lung metastasis mouse model by intravenously injecting MKN45 cells into mice (Fig. 5e). In this model, tumor cells were detected in the lungs (Fig. 5f), successfully mimicking the lung metastasis of gastric cancer. One and two weeks after tumor cell implantation, the mice were infused with two doses of PBS, 1 × 107 CIML NK cells, untargeted anti-non CAR-like NK cells, or anti-MSLN CAR-like NK cells. The infused NK cells were supported by rh-IL2 (intraperitoneal injection), and tumor growth was monitored weekly via BLI (Fig. 5f). Overall, anti-MSLN CAR-like NK cells significantly suppressed tumor cells in the lungs of most mice, whereas CIML NK and anti-non CAR-like NK cells failed to effectively control tumor progression (Fig. 5f, g). In addition, treatment with anti-MSLN CAR-like NK cells was well tolerated, with body weights remaining stable across all groups (Supplementary Fig. S3d). These findings suggest that anti-MSLN CAR-like NK cells can safely and effectively inhibit the distant metastasis of MSLN-positive tumor cells.
Next, we assessed the antitumor efficacy and specificity of anti-MSLN CAR-like NK cells in a peritoneal disseminated tumor mouse model. MSLN-high-expressing tumor cells (MKN45 and NCI-N87) and MSLN-low-expressing tumor cells (MiaPaCa-2) were injected intraperitoneally into the mice (Fig. 5h). Ten days later, tumor cell proliferation in the peritoneum was detected via BLI. The mice were then divided into two experimental groups: one group received an intraperitoneal infusion of 1 × 10⁷ CIML NK cells (Ctrl NK group), and the other group was infused with 1 × 10⁷ anti-MSLN CAR-like NK cells. The results revealed that anti-MSLN CAR-like NK cells significantly inhibited the growth of MSLN-high-expressing tumor cells, whereas tumors in the control group progressed rapidly (Fig. 5i, k). BLI images revealed significant differences between the anti-MSLN CAR-like NK cell treatment group and the control group at days 7 and 14 (Fig. 5l, n). However, for mice bearing MiaPaCa-2 tumors with low MSLN expression, CAR-like NK cell therapy did not significantly slow tumor growth or disease progression (Fig. 5j, m). No toxic reactions, such as weight loss, were observed in the anti-MSLN CAR-like NK-treated mice with disseminated peritoneal tumors (Supplementary Fig. S3e). Together, our data demonstrate that anti-MSLN CAR-like NK cells exhibit specific and potent antitumor activity against MSLN-positive tumor cells in vivo.
CAR-like NK cells engineered for tumor penetration display superior homing and infiltrative capacity
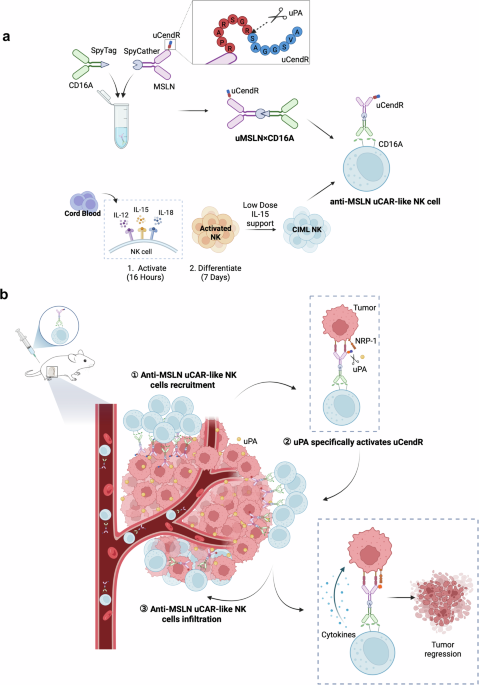
Although anti-MSLN CAR-like NK cells have demonstrated potent antitumor activity in vivo, the dense extracellular matrix and high interstitial pressure of solid tumor tissue often limit the infiltration capacity of immune cells, thereby compromising therapeutic efficacy.9 To enhance the penetration of anti-MSLN CAR-like NK cells into the tumor stroma and further improve antitumor efficacy, we conjugated the tumor-penetrating peptide uCendR (RPARSGR↓SAGGSVA)17 to the MSLN end. This modification, which is based on the common cleavage motif of urokinase-type plasminogen activator (uPA), endows anti-MSLN uCAR-like NK cells with both tumor-targeting and tissue-penetrating capabilities. Notably, uPA (PLAU) and NRP-1 transcripts are significantly upregulated across most cancer types (Supplementary Fig. S4a).
To investigate the tumor penetration of anti-MSLN uCAR-like NK cells in vitro, they were labeled with CFSE and coincubated with HGC27-MCSs for 6 h. As shown in Fig. 6a, CIML NK, anti-non CAR-like NK, and anti-MSLN CAR-like NK cells were confined to the periphery of the tumor spheroid, with only weak signals detected. In contrast, anti-MSLN uCAR-like NK cells actively penetrated the tumor spheroid, demonstrating significantly stronger penetration ability, which was 5.3 times greater than that of CIML NK cells, 5.8 times greater than that of anti-non CAR-like NK cells, and 3.2 times greater than that of anti-MSLN CAR-like NK cells (Fig. 6c).

Anti-MSLN uCAR-like NK cells demonstrate superior tumor infiltration and antitumor efficacy. a Confocal microscopy images (upper and middle panels) and surface plot images (lower panel) showing the penetration of CFSE-labeled CIML NK, anti-non CAR-like NK, anti-MSLN CAR-like NK, or anti-MSLN uCAR-like NK into HGC27-MCSs at an E:T ratio of 10:1. b Evaluation of the cytotoxicity of each group in (a) to HGC27-MCSs. c Mean CFSE fluorescence intensity of HGC27-MCS in (a). d Quantification of live/dead cells in HGC27-MCS from (b). e Representative in vivo images at different time points following intravenous injection of different cell types (CIML NK, anti-non CAR-like NK, NK + MSLN×CD16A, anti-MSLN CAR-like NK, or anti-MSLN uCAR-like NK) in MKN45 subcutaneous tumor-bearing mice, with tumor locations marked by circles. f Time-dependent changes in the signal intensity of different cell types in the tumors shown in (e). g Ex vivo images of the heart, liver, spleen, lung, kidney, and tumor 24 h after intravenous injection. h In the orthotopic pancreatic cancer mouse model, CIML NK cells, anti-MSLN CAR-like NK cells, or anti-MSLN uCAR-like NK cells (1 × 10⁷) were administered intraperitoneally, with treatment repeated twice at 7-day intervals. Recombinant human IL-2 was given concurrently. The tumor burden was assessed via BLI on days 0, 7, and 14. i Quantification of bioluminescent tumor images at different time points, as shown in (h). For c and d, one-way ANOVA and Tukey’s multiple comparison test were used. For f and i, two-way ANOVA and Tukey’s multiple comparison test were used. The data represent the means ± SEMs; n = 3 independent experiments. Scale bar: 100 µm. ns not significant; ∗P < 0.5; ∗∗P < 0.01; ∗∗∗P < 0.001; ∗∗∗∗P < 0.0001
In vivo experiments further validated the homing and penetration capabilities of anti-MSLN uCAR-like NK cells. In the subcutaneous tumor model of gastric cancer, anti-MSLN uCAR-like NK cells accumulated significantly at the tumor site, reaching a peak 24 h after treatment (Fig. 6e, f). In contrast, CIML NK and anti-non CAR-like NK cells presented minimal signals in the tumor region because of the lack of targeted localization. Notably, although MSLN×CD16A combined with CIML NK cells demonstrated a certain homing effect, the signals were weaker than those of anti-MSLN CAR-like NK cells assembled in vitro, potentially explaining why MSLN×CD16A combined with CIML NK cell therapy exhibited inferior antitumor efficacy in vivo. The tumors were resected and measured in vitro 24 h after infusion (Fig. 6g). Consistent with the results from whole-body imaging, the highest degree of cell accumulation at the tumor site was observed in the anti-MSLN uCAR-like NK group. Ex vivo organ imaging also revealed that the infused cells primarily accumulated in the liver and spleen, with minimal accumulation in other organs. The accumulation of anti-MSLN uCAR-like NK cells in non-tumor sites aligns with the natural homing and distribution patterns of NK cells, which are known to reside in the liver and spleen. In summary, anti-MSLN uCAR-like NK cells exhibited superior penetration and homing capabilities in MCSs and tumor tissues.
Given the superior penetration and homing properties of anti-MSLN uCAR-like NK cells, we further evaluated their antitumor efficacy. As expected, anti-MSLN uCAR-like NK cells exhibited superior antitumor efficacy (Supplementary Fig. S4b, c). Notably, anti-MSLN uCAR-like NK cells exhibited antitumor activity comparable to that of anti-MSLN CAR-like NK cells against MSLN-positive targets. This may be attributable to the absence of a penetration step during their interaction with monolayer tumor cells in vitro. We then evaluated anti-MSLN uCAR-like NK cells in the HGC27-MCS model, where their improved penetration translated into markedly enhanced tumor-targeted cytotoxicity (Fig. 6b, d). Anti-MSLN uCAR-like NK cells achieved approximately 80% cytotoxicity, which was significantly greater than that of anti-MSLN CAR-like NK cells (~50%, Fig. 6d). In the subcutaneous MKN45 (MSLN+) xenograft model, anti-MSLN uCAR-like NK cells significantly inhibited tumor growth with good tolerability (Supplementary Fig. S5). The adaptor protein used to arm anti-MSLN uCAR-like NK cells was incubated with human PBMCs for 24 h without target cells. No PBMC activation was observed, which is consistent with a favorable safety profile and a low risk of target-independent immune stimulation (Supplementary Fig. S4d). To better understand the effects of the tumor microenvironment and tumor barriers on immune cell penetration, we used a pancreatic orthotopic tumor model. In this model, MSLN-positive CFPAC-1 human pancreatic cancer cells were injected into the pancreas of mice. Three weeks after tumor cell injection, two doses of PBS, anti-MSLN CAR-like NK cells, or anti-MSLN uCAR-like NK cells (1.0 × 10⁷) were administered intraperitoneally, with rh-IL2 administered every other day. The PBS control group presented the most pronounced tumor invasion and disease progression. In contrast, anti-MSLN uCAR-like NK cells significantly inhibited tumor growth. Anti-MSLN CAR-like NK cells, which only exhibited targeted effects, tended to slow tumor growth and disease progression, but their efficacy was inferior to that of anti-MSLN uCAR-like NK cells with penetrating activity (Fig. 6h, i). Overall, our data demonstrate that anti-MSLN uCAR-like NK cells exhibit significant advantages in terms of antitumor efficacy.