
Informed consent and ethics committee approvals
All animal procedures followed the institutional laboratory animal research guidelines and were approved by the governmental authorities (Regional Administrative Authority Karlsruhe, Germany). Human tissues were used after approval of the local regulatory authorities (Ethics Committee at the Medical Faculties of the University of Heidelberg).
Mice
APP23-tg mice contain a human amyloid precursor protein (APP751) cDNA with the Swedish double mutation at position 670/671 under the control of the neuron-specific Thy-1 promoter (Calhoun et al., 1999). Heterozygote B6, D2-TgN[Thy-APPSWE]−23- tg mice (APP23) provided by Matthias Staufenbiel (Novartis Institutes for BioMedical Research, Novartis Pharma AG, Basel, Switzerland) were backcrossed twice with C57BL/6 mice (Janvier, Saint Berthevin Cedex, France) and kept under a 12/12 h light/dark cycle with standard food and water ad libitum. Wild-type (WT) littermates were used as control animals.
The Gt(ROSA)26Sor^tm1.1(CAG-cas9*,-EGFP) Fezh mouse line was acquired from Jackson Laboratory. It carries a Cas9-EGFP construct inserted into the Rosa26 locus, allowing ubiquitous expression of Cas9 and EGFP under the control of the CAG promoter. Mice were housed under specific pathogen-free conditions with a 12 h light/12 h dark cycle, at an ambient temperature of 22 ± 2 °C and relative humidity of 50–60%, with ad libitum access to food and water.
Human Alzheimer’s disease tissues
Cryo-conserved post-mortem Alzheimer’s disease patient tissue was obtained from the Neurobiobank Munich (Center for Neuropathology and Prion Research, Munich). All participants gave prospective pre-mortem written consent for their brains to be banked and used for research. Patient demographics and information on disease staging are summarized in Supplementary Table 3.
Immunofluorescence of formalin-fixed paraffin-embedded tissue
APP23-tg mice were perfused with 4% formalin (RotiHistofix 4%, Roth) under isoflurane anaesthesia. Brains were dissected and fixed for a maximum of 24 h in 4% formalin at 4 °C. Coronal tissue slides of the cortex were cut with a thickness of 7 µm using a microtome (Thermo Scientific). For immunofluorescence stainings, paraffin was removed by incubating the slides at 37 °C for 5 min and placing them into two cups of xylol for 10 min each. To rehydrate sections, they were transferred to two containers of 100% Ethanol for 5 min each, and a decreasing Ethanol-row (96%, 70%, and 50%) was performed for 3 min each. Slides were collected in VE water. For antigen retrieval, slides were transferred into a plastic cuvette, sealed with parafilm, and boiled in Cell Conditioning Solution pH 6.0 (Ventana, Roche Diagnostics) for 30 min in a food steamer. Sections were cooled down for 20 min in the same plastic cuvette and washed 3 times in washing buffer (0.1% Tween20 in 1x PBS). Immunofluorescence stainings were conducted sequentially, meaning that primary antibodies for different targets were not mixed but incubated one after another together with their matching secondary antibody.
The tissue was blocked with the appropriate serum, matching the species of the secondary antibody (4% goat or donkey serum in washing buffer) for 1 h at RT. Slides were incubated overnight at 4 °C with the respective primary antibody targeting CD3 (polyclonal rabbit anti-mouse/human, DAKO A0452, 1:200), CD31 (polyclonal rabbit anti-mouse, Abcam 28364, 1:100), or amyloid beta (monoclonal mouse anti-mouse, BioLegend 803014, 1:200) in blocking buffer. The next day, slides were washed 3 times with washing buffer before they were incubated with fluorescently labelled secondary antibodies to target CD3 (polyclonal goat anti-rabbit AF633, Thermofisher A-21070, 1:200), CD31 (polyclonal goat anti-rabbit AF546, Thermofisher A11010, 1:500), or amyloid beta (polyclonal goat anti-mouse AF488, Thermofisher A11029, 1:1000) for 1 h at RT. After thorough washing, sections were mounted using VECTASHIELD HardSet Mounting Medium with DAPI, which also stains Amyloid beta plaques. Images were acquired on an LSM700 confocal microscope (Zeiss) with 20x magnification.
Immunofluorescence of fixed frozen tissue
APP23-tg mice and Wt littermates were perfused with ice-cold PBS under ketamine/xylazine anaesthesia (120 mg/kg ketamine + 20 mg/kg xylazine in sterile 0.9% NaCl solution, 0.1 ml/10 g body weight). Brains were dissected and snap-frozen in Tissue-Tek® (Sakura). Coronal tissue slides of the cortex were cut with a thickness of 7 µm using a microtome (Thermo Scientific). For immunofluorescence stainings, the tissue was blocked with the appropriate serum, matching the species of the secondary antibody (4% goat or donkey serum in washing buffer) for 1 h at RT. Slides were incubated overnight at 4 °C with the respective primary antibody targeting CD3 (polyclonal rabbit anti-mouse/human, DAKO A0452, 1:200), CD31 (polyclonal rabbit anti-mouse, Abcam 28364, 1:100), or amyloid beta (monoclonal mouse anti-mouse, BioLegend 803014, 1:200) in blocking buffer. The next day, slides were washed 3 times with washing buffer before they were incubated with fluorescently labelled secondary antibodies to target CD3 (polyclonal goat anti-rabbit AF633, 1:200), CD31 (polyclonal goat anti-rabbit AF546, 1:500), or amyloid beta (polyclonal goat anti-mouse AF488, 1:1000) for 1 h at RT. After thorough washing, sections were mounted on day 4 using a mounting medium with DAPI. Images were acquired on an LSM700 confocal microscope (Zeiss) with 20x magnification.
T-cell quantification
T-cells were quantified at 8, 12, 16, 20, and 24 months of age in cortices of APP23tg mice. Formalin-fixed paraffin-embedded tissue was used, and T-cells were stained as previously described. Three mice per age and three brain slides per mouse were used. Three tile scan images of an area of 960.25 µm² were acquired for each slide, and T-cells were counted using an ImageJ macro. The background was subtracted from both the DAPI and Alexa Fluor 633 (AF633) channels, using the “Rolling Ball Background Subtraction” with a radius of 30 pixels. Subsequently, the images were filtered using the “Gaussian Blur” function with the radius set to 5. DAPI images were segmented using the “Find Maxima” tool with the prominence set to 1, and the threshold set accordingly. The output of Find Maxima was single points highlighting the centre of each DAPI-stained nucleus. The threshold for a true positive signal in the AF633 channel was set individually, and an area of the red signal was created. All single points marking DAPI maxima that were detected within the selected red area were counted as T-cells, whereas, all DAPI maxima were counted as total cells. For statistical analysis of T cell infiltration, mean values of each slide per time point were used (n = 9). Data were checked for normality using Prism’s built-in D’Agostino & Pearson test, and changes in T cell numbers normalized to total cells were calculated by One-way ANOVA and Tukey’s post hoc test. The significance level was set to 0.05.
Parenchymal and vascular amyloid beta plaques were counted manually on each tile scan, utilizing the “cell counter” function in Fiji. For statistical analysis of total plaque burden, mean values of each slide per time point were used (n = 9). Data were checked for normality using Prism’s built-in D’Agostino & Pearson test, and changes in plaque numbers were calculated by One-way ANOVA and Tukey’s post hoc test. The significance level was set to 0.05.
Spearman correlation analyses were run in Prism and plotted in linear regression models. Therefore, all values, independent of the age group, were used as single data points for correlation analysis. 8-month-old mice do not show any plaque burden, and 12-month-old mice only in very few slides. Since there is a baseline abundance of T-cells in the brain, these two-time points were excluded from the “T cell/plaque” analyses, as this would have led to a skewed picture. T-cells surrounding either parenchymal or vascular amyloid deposits were counted manually on a set of well-defined plaques of both types to prevent double counting of T-cells. Therefore, definite plaques were marked, and a circle with a radius of 90 pixels was drawn around the center. T cells falling into the defined area were counted, and ratios of T-cells per parenchymal and vascular amyloid deposits were calculated.
Isolation of brain-associated immune cells
APP23tg/APP23wt mice were sacrificed by intraperitoneal overdose of Ketamin/Rompun; brains were isolated without prior perfusion to capture a comprehensive representation of the immune microenvironment, including both resident brain and infiltrating immune cells, followed by digestion with 50 ug/ml Liberase (Sigma) at 37 °C for 8 min and mechanical dissociation through 100 and 70 µm strainers (Miltenyi) to generate a single cell suspension. Subsequently, Myelin removal was performed using a 30% continuous Percoll (GE Healthcare) gradient and centrifugation at 2700 rpm.
Fluorescence-activated cell sorting of mouse brain immune cells and library preparation for single-cell RNA- and VDJ-seq
After preparation of the single cell suspension, staining for Fluorescence-activated cell sorting (FACS) was performed. Murine cells were blocked with anti-mouse CD16/32 (0.5 μg per well, eBioscience) and stained for CD45 (BV510, Biolegend 103138), CD3 (APC, Biolegend 100236), and CD11b (FITC, Biolegend 101206). In addition to that, eFluor 780 fixable viability dye (eBioscience) was used according to the manufacturer’s protocol to exclude dead cells. Furthermore, for single-cell RNA sequencing, titrated amounts of TotalSeq C hashtag antibodies (TotalSeq™-C0301 anti-mouse Hashtag 1 Antibody, Biolegend 155861; TotalSeq™-C0302 anti-mouse Hashtag 2 Antibody, Biolegend 155863) were added to the cell-antibody mix in a total of 200 µl and incubated for 30 min on ice. Leukocytes were sorted for sorted for subsequent steps using a CD45+ s. Cells were sorted on BDAria II through a 100 µM nozzle and 4-way purity. Cells were sorted in 5 µl 0.04% BSA in PBS and kept on ice until processing. Single-cell capture, reverse transcription, and library preparation were carried out on the Chromium platform (10x Genomics) with the Single Cell 5ʹ reagent v2 kit (10x Genomics) according to the manufacturer’s protocol using 8000–23,000 cells as input per channel. Each pool of cells was tested for library quality, and library concentration was assessed. Each of the final libraries was paired-end sequenced (26 and 92 bp) on one Illumina NovaSeq 6000 S2 lane.
Single-cell transcriptomic analyses
Quality control and normalization
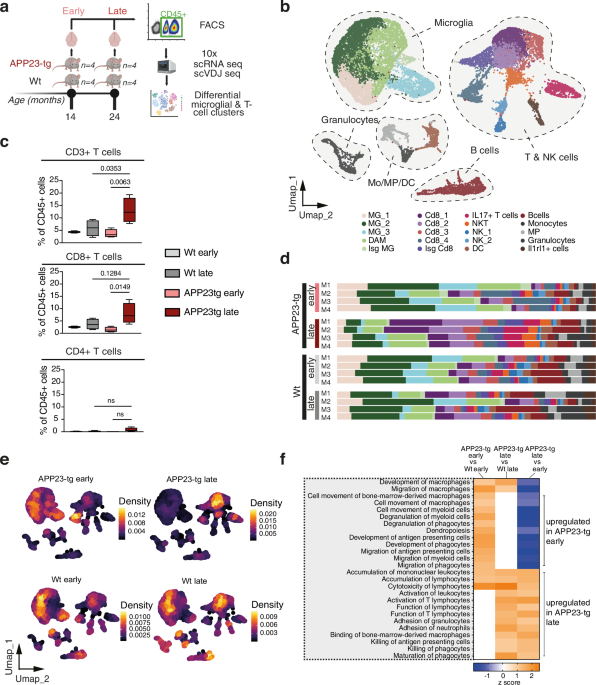
Single-cell RNA data were processed using the CellRanger pipeline (version 5.0) to the GRCm38 reference genome with all default settings. All downstream analyses were performed using Seurat v 4.4.0. All cells that had unique feature counts over 4000 (late time point)/3000 (early time point) or less than 200 (early and late time point), as well as cells with more than 5% mitochondrial counts, were excluded from downstream analyses. The cells of each mouse were stained with two different TotalSeq C hashtag antibodies during FACS staining. Using the HTODemux() command with default settings, following Seurat’s demultiplexing vignette (https://github.com/satijalab/seurat/blob/master/vignettes/hashing_vignette.Rmd), cells were demultiplexed to their respective sample of origin. Cells without an assigned hashtag (unassigned cells) or doublets (cells with two or more assigned hashtags) were removed from the analysis. Subsequently, only cells classified as singlets (67.8%) were used for further downstream analyses. Visualization of demultiplexing results, as well as unassigned cell/doublet proportions for each mouse, can be found in the Supplementary material section. Furthermore, mice of each group, as well as early and late time points, were merged, and a master object was created. Gene expression data were scaled and normalized using regularized negative binomial regression as implemented in ScTransform(). In addition to that, 2000 highly variable genes were selected also using ScTransform(). VDJ data from the filtered contig annotations files were added to the RNA data after running the cellranger 5.0 VDJ pipeline.
Integration of different groups and clustering
Subsequently, cells from all 16 mice were integrated using Seurat’s FindIntegrationAnchors() and IntegrateData() with standard parameters and ndims = 20. The data was scaled, and a principal component analysis was done using RunPCA() with npcs = 30. 20 principal components were selected based on the inflection point in the elbow plot. The generated dimensions were used to cluster cells by running FindNeighbours(), FindClusters(), and RunUMAP(). Umap was visualised using a resolution of 1.0. Differential gene expression analysis was performed using MAST (v1.26.0) to determine the identity of each cluster, and highly upregulated genes were used to label each cluster manually. To validate the cluster annotation, normalized expression values of 830 microarray samples of pure mouse immune cells from the Immunologic Genome Project (ImmGenData) were obtained using the celldex package (v1.10.0) and mapped onto the Umap using the SingleR package (v2.4.1).
Receptor-ligand interaction analysis
Receptor ligand analysis was performed using the cellchat package (v1.6.1). First, the dataset was split into four different groups (APP23-tg early/late, APP23-wt early/late), followed by creating cellchat objects for each of these groups using createCellChat() with default parameters. Transformed and normalized gene expression data were used as input, as well as the cluster annotation for defining the interacting cell groups. Subsequently, the CellChat database CellChatDB for murine analyses was obtained, containing 2021 validated molecular interactions. Overexpressed ligands and receptors were identified by running identifyOverExpressedGenes() and identifyOverExpressedInteractions(), and communication probability/strength between any interacting cell groups was calculated using computeCommunProb() as well as computeCommunProbPathway(). Lastly, to identify dominant senders, receivers, mediators, and influencers in all inferred communication networks netAnalysis_computeCentrality() was computed.
Ingenuity pathway analysis
QIAGENs Ingenuity Pathway Analysis (IPA) bioinformatic tool (accessed 2024) was used for downstream analyses and visualization of the scRNAseq dataset. The parent Seurat object was divided into individual objects consisting of single-cell clusters. For all CD45+ cells, differentially expressed (DE) genes between conditions (APP23tg versus Wt) or time points (late versus early) were identified by the FindMarkers() function that is based on a two-sided Wilcoxon rank-sum test. Average log2(fold change) values of DE genes were then supplied to IPA. To obtain heatmaps of “Diseases and functions” and “Canonical Pathways”, genes were filtered for mice as the species, immune cells as the tissue, and for a right-sided p-value < 0.05 by IPA’s implementation of Fisher’s exact test.
Single-cell T-cell receptor analysis
The T-cell receptor analysis was done using the scanpy (v1.10.1) and scirpy (v0.17.0) packages. The clustered data object from Seurat was loaded into a MuData object together with the receptor sequencing data. The data was subset to the CD8+ T-cells, and the clonal expansion was defined using the scirpy function tl.clonal_expansion() and analyzed for the subclusters. Differential gene expression analysis between the different clonal expansions was performed using the scanpy tl.rank_genes_groups() function.
Targeted spatial transcriptomics
Sample preparation
Coronal sections of the cerebral cortex of in total eight cryo-preserved APP23-tg and Wt mouse brains (n = 2 per group) were subjected to targeted spatial transcriptomics using the Molecular Cartography™ platform (Resolve Biosciences). For each mouse, a single brain hemisphere was analyzed. A coronally adjacent section was stained with pFTAA dye to detect Aβ deposits and guide ROI selection within the cerebral cortex for transcript detection. The list of transcripts probed is provided in Supplementary Table 1.
For human post-mortem Alzheimer’s disease patient tissue from the cerebral cortex, 1 cm3 cryo-conserved tissue cores were sectioned. Two sections per patient were used for spatial transcriptomics using the same platform as murine tissue. An immediately adjacent section was stained with Thioflavin S to detect Aβ deposits and guide ROI selection within the cerebral cortex for transcript detection. The list of transcripts probed is provided in Supplementary Table 2.
Methodology of Molecular Cartography platform
Molecular Cartography is an imaging-based, highly multiplexed single-molecule fluorescence in situ hybridization (smFISH) technology designed to detect and quantify individual RNA transcripts with subcellular resolution while preserving tissue morphology. The technique works in principle by employing a proprietary system of combinatorial barcoded probes and sequential imaging rounds. Briefly, custom designed, gene-specific oligonucleotide probes, each bearing a unique barcode, are hybridized to target RNA molecules within tissue sections. In subsequent iterative imaging cycles, these barcodes are decoded through the sequential binding and unbinding of fluorescent reporter molecules. Each cycle involves the detection of specific fluorophores associated with different barcode bits. By recording the fluorescent signal across multiple rounds, a unique “fingerprint” is generated for each RNA molecule, allowing its identification and precise spatial localization. This imaging-based method achieves subcellular resolution (approximately 300 nm), enabling the visualization of individual transcripts and their spatial context within cells and tissues. For our experiments, tissue sections were prepared according to Resolve Biosciences’ guidelines and processed in their service laboratory. Following data acquisition, transcripts were segmented to individual cells based on DAPI nuclear staining and Resolve Biosciences’ proprietary cell segmentation algorithm. Further information on the methodology is available on the company’s website as well as in the following publication63.
Mouse spatial transcriptomic analysis
For the cell segmentation-free analysis of the mouse spatial transcriptomics data, the raw data provided by the Molecular Cartography platform of Resolve Biosciences was loaded into a pandas dataframe. Additionally, the respective pFTAA stained images were imported using the tifffile package, normalized, and segmented via a threshold. Then the distance of each marker to the closest plaque was calculated using scipy.ndimage.distance_transform_edt(). The transcripts were then assigned to three categories, inside (d ≤ 0 µm), plaque adjacent (0 < d < =69 µm, corresponding to 500px on rendered images of assay slides), and outside (d > 69 µm). The colocalization of two markers was measured by individually calculating the histogram of the distance of marker 1 to a single transcript of marker 2. Then the histograms for all occurrences of marker 2 were averaged and renormalized by the annulus size of the respective bin π (bin2-(bin-1)2). To measure the colocalization of markers in tissue-adjacent regions, marker 2 occurrences were filtered to be only from the inside and plaque-adjacent regions.
The association of single markers with cell types and the colocalization of the marker in question were determined for a set of marker genes, where the colocalization was calculated for each marker gene (set as marker 2), and the mean was plotted. T-cell markers used were Cd3e, Cd4, Cd8b1, Foxp3, Trbc, Trdc, Cd163l1n and Ccr7; the microglia markers used were Cst3, Hexb, Tyrobp, Ctsd, P2ry12, Tmem119, Tgfbr1, Olfml3, Cst7, Apoe, Lpl, Trem2, Srgap2, and Nav2.
For the cell segmentation-based analysis of the mouse spatial transcriptomics data, segmented cells from the mouse samples were loaded in one scanpy AnnData object. The number of available markers varied across the samples. The markers were filtered to keep only markers with less than 60.000 missing values; the remaining missing values were set to 0. This resulted in a dataset with 97,596 cells and 109 markers. The cells were filtered to have a minimum of 20 counts expressed. Normalization was performed using Pearson residuals with sc.experimental.pp.normalize_pearson_residuals(), on the resulting residuals, the principal components were calculated and integrated with harmony (v0.0.10). The resulting integration was then clustered with Leiden clustering and visualized with a UMAP embedding (Fig. S6).
The gene panel was designed to differentiate cell types by including cell type markers, the marker sets for the individual cell types were scored for each cell with the sc.tl.score_genes() function (Fig. S6). Next, the Leiden clusters were assigned a cell type if the mean score for a cell type was significantly higher than in all other cells. Based on the following condition, for each Leiden cluster (lc) and cell type (ct), (mean(scorect (lc))-mean(scorect(other cells)))/(std(scorect(lc))+std(scorect(other cells)))>1, the clusters were annotated. This score uniquely assigned cell types to clusters, some clusters could not uniquely be annotated (Fig. S6).
Next, the data was subset to the T-cells, and PCA, harmony integration, and Leiden clusters were recomputed. To identify exhausted and ISG T-cells, the cells were scored for the respective gene sets: ISG genes: Ifit1, Ifit2, Irf7, Ifnar1, Usp18, Isg15, Cxcl10; exhaustion genes: Pdcd1, Havcr2, Lag3, Tigit, Ctla4, Cd244, Cd160, Cd96, Cd38, Tox. Based on these scores, two clusters were annotated as ISG T-cells, and one cluster was annotated as exhausted T-cells.
The distance of each cell to the plaques was calculated as previously described, and the histograms of the annotated cell types with respect to the distance from the plaque were plotted. The values of the histograms were renormalized to respect the quadratically increasing area covered by the bins.
Human spatial transcriptomics analysis
For the transcript-wise analysis of the spatial transcriptomics data, the raw output data describing transcript positions provided by the Molecular Cartography platform of Resolve Biosciences was loaded into a pandas dataframe. Additionally, the respective Thioflavin S-stained images were imported using the tifffile package, normalized, and segmented via a threshold. Then the distance d of the transcripts to the closest plaque was calculated using the scipy.ndimage.distance_transform_edt() function. The transcripts were then assigned to one of three categories, inside the plaque (d ≤ 0 µm), plaque adjacent (0 < d ≤ 69 µm, corresponding to 500px on rendered images of assay slides), and outside (d > 69 µm). Then the transcript density for each marker and each of the three categories was calculated by normalizing the number of transcripts with the size of the region. This transcript density was then plotted for each marker for the three regions.
Furthermore, histograms of the transcript density surrounding the plaques were generated with numpy.histogram(density=True). The histogram values were then renormalized by the annulus size of the respective bin π (bin2-(bin-1)2). The density histograms were generated per gene and compared to the histogram of all genes within the panel to capture relative overrepresentation.
Transwell migration assays
Isolation of murine T cells
Spleens were dissociated through 70 µm strainers into cold PBS. Erythrocytes were removed using a gentle hypotonic lysis adapted for Cas9/dCas9 mice (1 mL VE H₂O, 7 mL RPMI1640 medium, layered over 1 mL FBS, centrifuged 500 × g, 5 min). Cell suspensions were washed, filtered, and centrifuged (400 × g, 5 min, 4 °C). Splenocytes were resuspended in PBS and used for downstream applications.
CRISPR-based CXCR3 knock-out in murine T-cells
A PMSC-U6-sgRNA_scaffold-PGK-PuroR-BFP plasmid was used as the backbone for cloning single guide RNAs (sgRNAs) via Golden Gate assembly using BbsI. Three sgRNAs targeting Cxcr3 were designed using the VBC Score tool (https://www.vbc-score.org/), and the leading candidate was chosen for downstream experiments. A non-targeting sgRNA was included as a negative control (sequences provided in Supplementary Information). Retroviral particles were produced by transfecting Plat-E packaging cells with CXCR3-targeting or non-targeting (NT) gRNA constructs together with the helper plasmid pCL-Eco using Lipofectamine 2000 (Thermo Fisher Scientific). Viral supernatants were collected 48 h post-transfection, filtered through 0.45 µm filters, and used immediately for transduction. CD3⁺ T cells were isolated from spleens of Rosa26-Cas9 mice using mouse Pan T-cell isolation kit l (Miltenyi Biotec) according the manufacturer protocol. Cells were activated for 24 h in 24-well plates pre-coated with anti-CD3ε (1 µg/ml) and addition of anti-CD28 (2 µg/ml) and 100 U/ml recombinant human IL-2 at a cell density of 3 × 106cells/ml. RetroNectin-coated 24-well plates (25 µg/ml, Takara) were preincubated with PBS containing 2% BSA, rinsed, and loaded with viral supernatant. Plates were centrifuged at 2000 × g for 2 h at 32 °C, after which 1 × 106 activated T cells were added per well and spinoculated at 1500 rpm for 10 min. Cells were cultured for 4 days at 37 °C in IL-2–supplemented lymphocyte medium and split when necessary. Transduction efficiency was assessed by BFP⁺ and GFP⁺ fluorescence from the retroviral constructs and Rosa26-Cas9 mouse line, respectively. CXCR3 knockout was confirmed by flow cytometry. NT-transduced cells (NT KO) served as controls in all downstream assays.
CXCL10-mediated migration and CXCR3 blocking assay
For transwell migration assays, Millicell® hanging inserts (pore size 5 µm, Merck) were used. Recombinant mouse CXCL10 (200 ng/ml in serum-free RPMI) was added to the basolateral side (lower chamber) in a total volume of 600 µl. 1 × 106 CXCR3 KO or NT KO T-cells were added in a volume of 200 µl to the apical side (upper insert) in serum-free RPMI1640. Where indicated, a blocking anti-CXCR3 antibody (1 µg/ml, Biolegend, Clone:CXCR3-173) was included in the upper chamber. Cells were incubated for 8 h at 37 °C. Migrated cells in the lower chamber were collected, stained for CD3(PercP Cy5.5, Biolegend 100218), CD8(PE-Cy7, Biolegend 100722), and CXCR3 (APC, Biolegend 126512), and analyzed by flow cytometry (ZE5 Cell analyzer Biorad). eFluor 780 fixable viability dye (Invitrogen) was used according to the manufacturer’s protocol to exclude dead cells, and counting beads (123count eBeads, Invitrogen) were used to quantify absolute counts of migrated cells. GFP and BFP fluorescence were used to distinguish Cas9-expressing migrating cells and transduced populations, respectively.
ISG induction and ISG-T cell mediated migration assays
CD8⁺ T cells were isolated from spleens of Rosa26-Cas9−/− mice (GFP-) using mouse CD8a+ T-cell isolation kit (Miltenyi Biotec) for isolating untouched CD8 T-cells according to the manufacturer’s instructions. Cells were activated for 24 h in 24-well plates pre-coated with anti-CD3ε (1 µg/ml) and addition of anti-CD28 (2 µg/ml) and 100 U/ml recombinant human IL-2 at a cell density of 3 × 106 cells/ml. Activated CD8⁺ T cells were harvested, washed in TexMACS medium (serum-free) to remove residual FBS, and seeded in the basolateral side of transwell plates (0.5 × 106 cells per well) in TexMACS medium.
ISG induction was achieved by treating CD8 + T cells in the basolateral well with the murine STING agonist DMXAA (10 µg/ml, Sigma-Aldrich) for 5 h at 37 °C. Control wells received vehicle (TexMACS medium with 0.1% DMSO). Similar to the CXCL10-mediated migration assay, 1 × 106 transduced CXCR3 KO and NT KO T-cells were added in a volume of 200 µl to the apical side in serum-free RPMI and assay incubated for 8 h at 37 °C. Number of migrated cells was assessed as above.
Data visualization
Data visualization was done using GraphPad Prism (v10). Visualization of single-cell data was performed using Seurat (v 4.4.0) with additional customization using ggplot2 (v3.5.1) where required. Graph types and statistical annotations are described in the corresponding figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.