
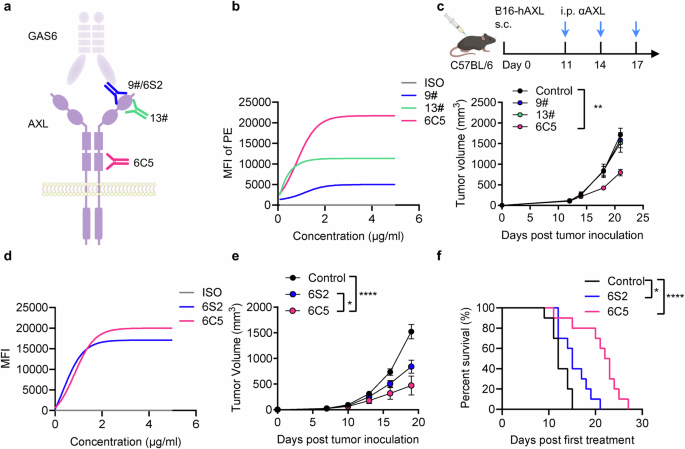
An antibody targeting the membrane-proximal epitope of AXL has enhanced antitumor efficacy
To explore the therapeutic potential of antibodies targeting distinct AXL epitopes, we developed several monoclonal antibodies against human AXL (hAXL). Clones 9# and 13# were found to bind the first Ig-like domain (domain 1), corresponding to membrane-distal epitopes. Clone 9# competes with the ligand GAS6 for binding to AXL, whereas clone 13# does not.26 In contrast, clone 6C5 binds to the second FNIII domain (domain 4), which represents a membrane-proximal epitope (Fig. 1a, Supplementary Fig. 1a, b). Compared with clones 9# and 13#, 6C5 exhibited stronger binding to hAXL on the cell membrane surface (Fig. 1b). Furthermore, we demonstrated that 6C5 binds to native AXL on primary human myeloid cells (Supplementary Fig. 1c). To evaluate the in vivo therapeutic efficacy of these antibodies, we engineered a B16-hAXL melanoma cell line by stably expressing hAXL in B16F10 mouse melanoma cells (Supplementary Fig. 1d). In a syngeneic mouse model, treatment with 6C5 exhibited significant therapeutic efficacy against subcutaneous B16-hAXL melanoma in both female and male mice (Fig. 1c and Supplementary Fig. 1e, f). Immunohistochemical analysis revealed no apparent histopathological abnormalities in major organs following 6C5 treatment (Supplementary Fig. 1g), and serum cytokine levels remained comparable to those in control mice (Supplementary Fig. 1h), indicating that 6C5 treatment did not induce systemic toxicity. Furthermore, 6C5 also potently inhibited the growth of CT26-hAXL colon tumors in BALB/c mice (Supplementary Fig. 1i), supporting its broad antitumor activity across different tumor types. We also tested the YW327.6S2 (6S2) antibody, developed by Genentech, which targets the first Ig-like domain and competes with GAS6 for AXL binding23 (Fig. 1a). 6S2 and 6C5 exhibited comparable binding affinities for hAXL (Fig. 1d). Notably, in vivo, compared with 6S2, 6C5 showed significantly superior antitumor efficacy in the syngeneic melanoma model (Fig. 1e, f). These results suggest that targeting the membrane-proximal epitope of AXL with 6C5 confers enhanced therapeutic benefit over the use of antibodies directed against membrane-distal domains.

An antibody targeting the membrane-proximal epitope of AXL has enhanced antitumor efficacy. a Schematic depiction of antibodies that bind to different epitopes of AXL. b Comparison of the binding affinities of anti-AXL antibodies for AXL on the cell surface. c Tumor growth curves of B16-hAXL-bearing female C57BL/6 mice (n = 5) treated with PBS (control), 9#, 13#, or 6C5 by intraperitoneal injection on days 11, 14, and 17. d Comparison of the binding affinities of 6S2 and 6C5 to AXL on the cell surface. Tumor growth curves (e) and survival curves (f) of B16-hAXL-bearing C57BL/6 mice (n = 10) treated with PBS (control), 6S2 or 6C5 cells by intraperitoneal injection on days 8, 11, 14, and 17. The data are presented as the means ± SEMs of two or three independent experiments. Statistical analysis of tumor growth was performed via two-way ANOVA. For the survival curve data in (f), the log-rank test was applied. ns (not significant), *P < 0.05, **P < 0.01 and ****P < 0.0001
The therapeutic efficacy of 6C5 is dependent on its binding to AXL on tumor cells
Inoculation of murine tumor cells expressing hAXL into wild-type mice may trigger xenogeneic immunogenic responses, potentially confounding the evaluation of antibody-based therapies. To enable a more accurate assessment, we generated hAXL-transgenic C57BL/6 mice (hAXLBAC mice) expressing hAXL (Supplementary Fig. 2a). Flow cytometric analysis of splenocytes confirmed hAXL expression predominantly in CD11c+ cells (Fig. 2a and Supplementary Fig. 2b), mimicking the expression pattern of AXL in human immune compartments and thereby increasing the translational relevance of the model. Using these transgenic mice, we compared the therapeutic efficacy of the 6C5 and 6S2 antibodies. 6C5 consistently outperformed 6S2 in suppressing B16-hAXL tumor growth (Fig. 2b). To further evaluate the cross-tumor applicability of 6C5 in hAXLBAC mice, we engineered an MC38-hAXL colon tumor cell line (Supplementary Fig. 2c). In hAXLBAC mice, compared with 6S2, 6C5 also demonstrated enhanced efficacy in inhibiting the growth of MC38-hAXL tumors (Fig. 2c). Given that most cancer-related deaths result from metastasis, we next explored the ability of 6C5 to control metastatic disease. In a lung metastasis model established by intravenous injection of B16-hAXL cells, 6C5-treated mice exhibited a marked reduction in metastatic nodules compared to the control group (Fig. 2d), indicating robust inhibition of metastatic tumor growth. Finally, to explore its translational potential in human cancers, we tested a chimeric 6C5 antibody version in humanized mouse models. The chimeric 6C5 effectively suppressed the growth of human pancreatic ductal adenocarcinoma CFPAC-1 tumors (Fig. 2e).

The therapeutic effect of 6C5 is dependent on its binding to AXL on tumor cells. a Flow cytometry was conducted to evaluate the expression of hAXL in splenocytes isolated from hAXL-transgenic mice (hAXLBAC mice). b Tumor growth curves of B16-hAXL-bearing hAXL-transgenic mice (n = 5) treated with PBS (control), 6C5, or 6S2 by intraperitoneal injection on days 8, 11, and 14. c Tumor growth curves of MC38-hAXL-bearing hAXL-transgenic mice (n = 5) treated with PBS (control), 6C5, or 6S2 by intraperitoneal injection on days 8, 11, and 14. d C57BL/6J mice (n = 5) were intravenously inoculated with 2 × 10⁶ B16-hAXL cells. Tumor-bearing mice received intraperitoneal injections of 6C5 (200 µg per mouse) on days 7, 10, 13, and 16 post-inoculation. Lungs were collected on day 18 following the first administration for tumor metastasis analysis. e NSG-SGM3 mice (n = 6) were inoculated with 1 × 10⁶ CFPAC-1 cells. On day 8 post-inoculation, the tumor-bearing mice received an intravenous injection of 1 × 10⁷ human PBMCs, followed by the intraperitoneal administration of PBS (control) or 6C5-hIgG1 (200 µg per mouse) on day 10 and treatment every two days. f Tumor growth curves of B16-hAXL-bearing Axl−/− C57BL/6 mice (n = 6) treated with PBS (control) or 6C5 by intraperitoneal injection on days 8, 11, and 14. g Tumor growth curves of B16F10- or B16-hAXL-bearing C57BL/6 mice (n = 6) treated with PBS (control) or 6C5 by intraperitoneal injection on days 11, 14, and 17. h Tumor growth curves of B16-hAXL-bearing Rag1−/− mice treated with PBS (control, n = 4) or 6C5 (n = 5) by intraperitoneal injection on days 9, 12, and 15. The data are presented as the means ± SEMs, and two or three independent experiments were performed. Statistical analysis of tumor growth was performed via two-way ANOVA. For (d), unpaired two-tailed t-tests were applied. ns (not significant), *P < 0.05, **P < 0.01, ***P < 0.001
Because AXL is also present on host cells, such as M2 macrophages6,27 and DCs,28 we investigated whether the efficacy of 6C5 involves targeting the host’s AXL. 6C5 was found to bind murine AXL (mAXL) but with weak affinity (Supplementary Fig. 2d). The antibody retained antitumor efficacy in Axl-knockout mice, indicating that host AXL expression is not necessary (Fig. 2f). In contrast, 6C5 had no therapeutic benefit in tumors lacking hAXL, highlighting the necessity of AXL expression on tumor cells (Fig. 2g). The activation of AXL signaling has been demonstrated to promote tumor progression.29 We next examined whether 6C5 exerts a direct effect on tumor cell proliferation. In vitro treatment of B16-hAXL melanoma cells with 6C5 showed no growth inhibition (Supplementary Fig. 2e). Similar results were observed in the AXL-high human tumor cell lines SN12C and Calu-1; only cabozantinib, a tyrosine kinase inhibitor, significantly inhibited proliferation, whereas 6C5 did not (Supplementary Fig. 2f, g). Moreover, in vitro treatment with these AXL antibodies increased the phosphorylation of AXL and its downstream Akt effector molecules in cancer cells, suggesting that they do not block but rather activate AXL signaling (Supplementary Fig. 2h). Interestingly, both the 6C5 antibody and its counterpart antibody 1# led to a decrease in the classical AXL band at approximately 140 kDa along with a concurrent increase in the lower 100 kDa molecular band, suggesting a possible modification change or truncation of AXL molecules after treatment with these antibodies (Supplementary Fig. 2h). These findings suggest that the efficacy of the 6C5 antibody depends on its interaction with AXL on tumor cells, but it does not directly affect tumor growth through the modulation of AXL signaling.
Given the lack of direct cytotoxicity, we hypothesized that 6C5 mediates tumor control via immune mechanisms. In support of this finding, 6C5 failed to control B16-hAXL tumor growth in immunodeficient Rag1−/− and NSG-SGM3 mice, which lack functional T and B cells (Fig. 2h and Supplementary Fig. 2i). These data demonstrate that the therapeutic efficacy of 6C5 requires adaptive immunity and is dependent on AXL expression on tumor cells.
AXL Ab treatment modulates the TME to increase TIICs infiltration and antitumor CD8+ T immunity
Given that adaptive immunity is required for AXL Ab-mediated tumor control, we further investigated changes in tumor-infiltrating immune cells (TIICs). We sorted intratumoral CD45+ cells from treated tumors and conducted single-cell sequencing to analyze their phenotypic and transcriptional landscape. Unsupervised clustering identified 22 distinct immune cell clusters, which were visualized via UMAP (Supplementary Fig. 3a). Macrophages and monocytes (clusters 0, 1, 2, 4, 5, 6, 7, 8, and 10) constitute the majority of TIICs (Supplementary Fig. 3a, b). Moreover, substantial upregulation of distinct chemokine profiles was observed in TAMs following 6C5 treatment (Fig. 3a). These findings suggest that 6C5 promotes a proinflammatory myeloid phenotype, which may enhance immune cell infiltration. To confirm this, we next performed flow cytometric analysis of TIICs. Indeed, following 6C5 treatment, multiple myeloid and lymphoid populations, including DCs, monocytes, neutrophils, T cells, and B cells, were increased within the tumor (Fig. 3b, c), indicating effective remodeling of the TME toward a more immunologically “hot” phenotype. Consistently, immunofluorescence analysis of intratumoral CD45+ leukocytes and CD8+ T cells further confirmed that 6C5 treatment markedly enhanced immune cell infiltration within the tumor (Supplementary Fig. 3c).

AXL Ab treatment modulates the TME to increase TIIC infiltration and antitumor CD8+ T immunity. a Heatmap showing unsupervised hierarchical clustering of chemokine gene expression profiles in macrophage populations. The number of intratumoral myeloid (b) and lymphoid immune (c) cells was analyzed on day 17 in B16-hAXL-bearing C57BL/6 mice that were treated intraperitoneally with 6C5 on days 12 and 15. d Uniform manifold approximation and projection (UMAP) analysis of total CD8+ T cells in tumors. e FeaturePlot visualization of effector molecule expression in intratumoral CD8+ T cells from the control and 6C5-treated groups. f Dot plot of genes associated with T-cell proliferation, metabolism, costimulatory signaling, and transcriptional regulation in Cluster 3 CD8+ T cells from the control and 6C5-treated groups. The frequency (g) and quantity (h) of intratumoral 4-1BB+CD8+ T cells were analyzed on day 15 in B16-hAXL-bearing C57BL/6 mice that were treated intraperitoneally with 6C5 on days 11 and 14. i Tumor growth curves of B16-hAXL-bearing C57BL/6 mice (n = 5) treated with PBS (control), 6C5 or 6C5 combined with an anti-CD8 antibody (αCD8). j Tumor growth curves of B16-hAXL-bearing C57BL/6 mice (n = 10) treated with PBS (control), 6C5 or 6C5 combined with an anti-CD4 antibody (αCD4). The data are presented as the means ± SEMs, and two or three independent experiments were performed. Statistical analysis of tumor growth was performed via two-way ANOVA. For (b, c, g, h), unpaired two-tailed t-tests were applied. ns not significant, *P < 0.05, **P < 0.01, and ***P < 0.001
Given the critical role of CD8+ T cells in antitumor immunity, we further classified CD3+CD8+ T cells from total immune cells. Five distinct subpopulations (C1–C5) were identified and visualized via UMAP for dimensionality reduction (Fig. 3d and Supplementary Fig. 3d). Among the CD8+ T-cell subsets, Cluster 3 (C3) was distinguished by high expression of effector genes (Gzmb, Prf1 and Ifng), indicating a cytotoxic function (Fig. 3e). Notably, 6C5 treatment increased both the abundance and effector activity of this cluster within the CD8+ T-cell compartment (Fig. 3e and Supplementary Fig. 3e). Additionally, 6C5 treatment upregulated genes associated with T-cell proliferation (Top2a, Pcna, and Cdk2), metabolism (Mtor, Hif1a, Slc2a1, Stat3, and Stat5b), costimulatory signaling (Cd28, Icos, Tnfrsf9, Tnfrsf14, and Tnfrsf18), and transcription regulation (Tbx21, Eomes, Runx3, and Bach2) in C3 (Fig. 3f). This population expressed high levels of 4-1BB (Tnfrsf9) and was thus designated 4-1BB+CD8+ T cells (Supplementary Fig. 3f). We further analyzed the impact of 6C5 treatment on intratumoral 4-1BB+CD8+ T cells via flow cytometry (Supplementary Fig. 3g). Flow cytometric analysis confirmed a substantial increase in both the frequency and number of these effector CD8+ T cells after 6C5 treatment (Fig. 3g, h). To directly assess their role, we depleted CD8+ T cells during therapy. CD8+ T-cell depletion completely abolished the antitumor activity of 6C5, confirming its essential role in mediating therapeutic efficacy (Fig. 3i and Supplementary Fig. 3h). While CD8+ T cells are typically supported by CD4+ T cells,30,31 we surprisingly found that depleting CD4+ T cells enhanced 6C5 efficacy (Fig. 3j and Supplementary Fig. 3i), suggesting the induction of an immunosuppressive CD4+ T-cell subset following 6C5 treatment.
Membrane-proximal-targeting AXL Ab activates APCs for tumor control with potent ADCP induction ability
Considering the critical role of CD8+ T cells in mediating effective anti-AXL therapy, we next investigated the mechanisms by which anti-AXL enhances T-cell responses. Engagement of activated Fc receptors (FcγRs) on APCs via the antibody Fc domain could induce phagocytosis of target tumor cells and subsequent cross-presentation of tumor antigens to activate CD8+ T cells. In addition, Fc-mediated effector functions may trigger ADCC through natural killer (NK) cells to kill tumor cells. To delineate the immune effector mechanisms involved, we performed selective immune cell depletion experiments. While depletion of NK cells did not affect tumor control, the therapeutic efficacy of the 6C5 antibody was completely abolished after macrophage depletion, indicating that macrophages, but not NK cells, are essential mediators of 6C5-induced tumor control (Fig. 4a–c and Supplementary Fig. 4a, b). Next, we investigated the impact of the 6C5 antibody on the phagocytosis of tumor cell antigens by macrophages. Tumor cells were labeled with CFSE and subsequently incubated with macrophages. Flow cytometry analysis revealed that the addition of the 6C5 antibody significantly enhanced the phagocytic ability of the macrophages (Fig. 4d and Supplementary Fig. 4c). In contrast, the Fc variant of 6C5 that lacks the ability to bind FcγR exhibited minimal phagocytic activity (Fig. 4d and Supplementary Fig. 4c) and completely lost its antitumor efficacy (Fig. 4e). These results illustrate the critical role of Fc-dependent macrophage engagement in AXL Ab-mediated tumor control.

Membrane-proximal targeting of the AXL Ab activates APCs for tumor control with potent ADCP induction ability. a Workflow for B16-hAXL tumor modeling with 6C5 + αNK1.1 or αCSF1R treatment. Tumor growth curves (b) and survival curves (c) of B16-hAXL-bearing C57BL/6 mice treated with PBS (control, n = 14), 6C5 (n = 14), αNK1.1 (n = 7), αCSF1R (n = 7), 6C5 + αNK1.1 (n = 6), or 6C5 + αCSF1R (n = 7). d The frequency of CFSE+ macrophages was analyzed 2 h after coculture with B16-hAXL tumor cells and macrophages in vitro, both in the presence and absence of 6C5, Fc mutated variant 6C5 (6C5-mutant), or an isotype control (Iso). e Tumor growth curves of B16-hAXL-bearing C57BL/6 mice treated with PBS (control, n = 6), 6C5 (n = 6) or the 6C5-mutant (n = 5). f The frequency of CFSE+ macrophages was analyzed 2 h after coculture with B16-hAXL tumor cells and macrophages in vitro, both in the presence and absence of the indicated antibodies. g Tumor growth curves of B16-hAXL-bearing CD11c-DTR C57BL/6 mice treated with control (PBS, n = 6), 6C5 (n = 7), or 6C5 + DT (n = 7) are shown. The data are presented as the means ± SEMs, and two or three independent experiments were performed. Statistical analysis of tumor growth was performed via two-way ANOVA. For the survival curve data in (c), the log-rank test was applied. ns (not significant), *P < 0.05, **P < 0.01 and ****P < 0.0001
We hypothesized that variations in the abilities of different anti-AXL monoclonal antibodies to induce ADCP activity may be attributed to their different epitope targeting abilities, ultimately leading to distinct therapeutic efficacy. To test this hypothesis, we compared several anti-AXL antibodies that target either membrane-proximal (the second FNIII domain) or membrane-distal epitopes (the first Ig-like domain) (Supplementary Fig. 1a, b). Antibodies (1#, 20#, and 22#) targeting the membrane-proximal epitopes of AXL potently induced macrophage-mediated phagocytosis of tumor antigens, demonstrating an efficiency comparable to that of 6C5 and surpassing the effectiveness of antibodies (9#, 13#, and 6S2) targeting membrane-distal epitopes (Fig. 4f and Supplementary Fig. 4d). Moreover, in vivo treatment with antibodies (1#, 20#) targeting the membrane-proximal epitopes significantly inhibited the growth of B16-hAXL melanoma (Supplementary Fig. 4e), indicating that the increased ADCP induced by the binding of antibodies to the membrane-proximal epitopes of AXL is associated with improved therapeutic outcomes.
Finally, whether macrophages can directly function as APCs to activate CD8+ T cells or require the involvement of DCs remains unclear. To address this question, we treated Batf3−/− mice, which specifically lack type 1 conventional dendritic cells (cDC1s), with a 6C5 antibody and observed a decrease in antitumor efficacy (Supplementary Fig. 4f). Similarly, depletion of DC cells by diphtheria toxin (DT) impaired AXL antibody-mediated tumor control in CD11c-DTR mice (Fig. 4g). Collectively, these findings indicate that the therapeutic efficacy of 6C5 is dependent on APCs, including both macrophages and DCs.
AXL Ab treatment activates type I interferon signaling in APCs to trigger antitumor immunity
Type I interferons are essential for the cross-priming of CD8+ T cells by APCs.32 Single-cell RNA-sequencing of tumors treated with 6C5 revealed broad upregulation of interferon-stimulated genes (ISGs) in both the macrophage and DC subsets (Fig. 5a). Further analysis revealed that IFN-β was the dominant subtype expressed, primarily by monocytes and macrophages (Fig. 5b and Supplementary Fig. 5a), whereas macrophages (clusters 0, 1, 2, 4, 6, 7, and 10), characterized by the expression of Adgre1, Cd68, and Fcgr1, constituted the majority of intratumoral immune cells (Fig. 5b and Supplementary Fig. 3b). These results identify macrophages as the principal intratumoral source of IFN-I. To investigate the effect of antibodies on IFN-β production by macrophages, we co-incubated macrophages with tumor cells in vitro with or without antibodies. After a 24-h incubation, the 6C5 antibody significantly upregulated IFN-β production in the incubation system (Fig. 5c). In contrast, the addition of an Fc-silencing 6C5 antibody did not increase IFN-β production compared with that in the control group (Fig. 5c). These findings collectively show that 6C5 promotes macrophage-mediated phagocytosis and IFN-I secretion.

AXL Ab treatment activates type I interferon signaling in APCs to trigger antitumor immunity. a Violin plots showing the expression levels of interferon-stimulated genes (ISGs) across different DC and macrophage subsets. b Feature plot showing the expression of Ifnb1 in immune cells. c IFN-β in the supernatant was analyzed in macrophages and the B16-hAXL tumor cell coculture system in the presence and absence of the indicated antibodies. d Tumor growth curves of B16-hAXL-bearing C57BL/6 mice (n = 5) treated with PBS (control), 6C5, or 6C5 + αIFNAR1. e Tumor growth curves of B16-hAXL-bearing Tmem173−/− mice treated with PBS (control, n = 5) or 6C5 (n = 6). f Tumor growth curves of B16-hAXL-bearing Myd88−/− mice treated with PBS (control, n = 5) or 6C5 cells (n = 6). g Heatmap showing changes in the expression of antigen presentation- and migration-related genes in macrophage and dendritic cell (DC) subsets between the 6C5-treated and control groups. h Tumor growth curves of B16-hAXL-bearing C57BL/6 mice (n = 5) and Zbtb46-cre Ifnar1f/f C57BL/6 mice (n = 5) treated with PBS (control) or 6C5. The data are presented as the means ± SEMs, and two or three independent experiments were performed. Statistical analysis of the tumor growth curves was performed via two-way ANOVA. For (c), an unpaired two-tailed t-test was applied. ns not significant, **P < 0.01, ***P < 0.001, and ****P < 0.0001
To determine whether IFN-I induction is essential for tumor control, we used Ifnar1−/− mice. Indeed, 6C5 Ab treatment failed to inhibit tumor growth in Ifnar1−/− mice (Supplementary Fig. 5b). Similarly, blocking the IFN-I pathway in WT mice with an antibody impaired 6C5 antibody-mediated tumor control (Fig. 5d), suggesting that the IFN-I pathway is critical for mediating tumor control. IFN-I can be derived from the activation of the STING (Tmem173) or MyD88 (Myd88) pathways.33,34 In Tmem173−/− mice, the therapeutic efficacy of the 6C5 antibody was maintained (Fig. 5e). Conversely, the therapeutic effect of the antibody was entirely abolished in Myd88−/− mice (Fig. 5f). These results indicate that the 6C5 antibody induces IFN-I secretion via the MyD88 pathway for tumor control.
The activation of IFNAR signaling by IFN-I could promote the activation and antigen presentation of APCs, which further activate the T-cell response. Our single-cell RNA-sequencing data revealed that treatment with 6C5 led to the upregulation of several antigen presentation-related costimulatory molecules (Cd40, Cd80, Cd86, and Cd83), MHC molecules (H2-Eb1, H2-Eb2, H2-K1, and H2-D1), and migration markers (Icam1 and Ccr7) across several macrophage and DC subsets (Fig. 5g). These transcriptional changes suggest that 6C5 promote APC cell activation, increase their antigen-presenting capacity and their migratory potential. Next, we investigated which cell subset expressing IFNAR is required for tumor control during 6C5 antibody treatment. We crossed the Ifnar1f/f allele35 onto the Zbtb46-cre and Lyz2-cre deletion strains36,37. The Zbtb46-cre Ifnar1f/f strain specifically deleted Ifnar1 within cDCs, whereas the Lyz2-cre Ifnar1f/f strain selectively deleted Ifnar1 in macrophages. Notably, 6C5-mediated tumor control was significantly impaired in Zbtb46-cre Ifnar1f/f mice (Fig. 5h), whereas 6C5 treatment was effective in Lyz2-cre Ifnar1f/f mice (Supplementary Fig. 5c). These findings demonstrate that the response of cDCs to IFN-I is essential for 6C5 treatment-induced antitumor immunity.
The 6C5 antibody overcomes melanoma resistance to immune checkpoint inhibitors
While 6C5 monotherapy resulted in significant tumor suppression, it failed to achieve complete tumor eradication. Investigations into resistance mechanisms revealed substantial loss of AXL expression in tumor cells during progression, with a more pronounced trend observed in animals treated with 6C5 (Supplementary Fig. 5d). These findings highlight the need for combination therapies to achieve durable therapeutic responses.
CD4+ T cells expanded during 6C5 treatment and appeared to exert immunosuppressive effects (Fig. 3c, j). Tregs are typically considered the major suppressive CD4⁺ T subset. However, combining the 6C5 Ab with an anti-CTLA-4 antibody failed to enhance therapeutic efficacy (Fig. 6a), suggesting that Tregs are not the primary mediators of immune suppression. The differences in gene expression patterns between Foxp3−CD4+ T cells from the 6C5-treated groups and those from the PBS-treated groups were positively correlated with the differences in gene expression between intratumoral Treg cells and Foxp3−CD4+ T cells from the PBS-treated group (Fig. 6b). These results suggested that intratumoral Foxp3−CD4+ T cells may adopt a transcriptional profile resembling that of Treg cells following 6C5 treatment. PD-1hiFoxp3−CD4+ T cells were previously shown to be induced in association with immunosuppression by anti-CTLA-4 antibody treatment in melanoma.38 Similarly, 6C5 treatment was associated with significant upregulation of PD-1hiFoxp3−CD4+ T cells within the tumor (Fig. 6c). We observed that global differences in gene expression between PD-1hi and PD-1−Foxp3−CD4+ T cells from the 6C5-treated group were positively correlated with global differences in gene expression between intratumoral Tregs and Foxp3−CD4+ T cells from the PBS-treated group (Fig. 6d). Consistently, gene set enrichment analysis (GSEA) of global gene expression differences between PD-1hi and PD-1−Foxp3−CD4+ T cells revealed an enrichment of genes upregulated in Treg cells compared with Foxp3−CD4+ T cells within the PD-1hi population (Fig. 6e), further suggesting that PD-1 expression is associated with Foxp3−CD4+ T cells enriched with a Treg cell–like transcriptional profile.

The 6C5 antibody overcomes melanoma resistance to immune checkpoint inhibitors. a Tumor growth curves of B16-hAXL-bearing C57BL/6 mice (n = 5) treated with PBS (control), 6C5, or 6C5 + αCTLA4. b Scatterplot comparing the global differences in gene expression between intratumoral Treg cells and Foxp3−CD4+ T cells from the PBS-treated group and the transcriptional differences between intratumoral Foxp3−CD4+ T cells treated with PBS and those treated with 6C5. c The number of intratumoral PD-1hiFoxp3−CD4+ T cells was analyzed in B16-hAXL-bearing C57BL/6 mice that were treated with PBS or 6C5. d Scatterplot comparing the global changes in gene expression between intratumoral Tregs and Foxp3−CD4+ T cells from the PBS-treated group and the transcriptional differences between intratumoral PD-1hi– and PD-1−Foxp3−CD4+ T cells from 6C5-treated mice. e GSEA demonstrating an enrichment of genes upregulated in Treg cells versus Foxp3−CD4+ T cells from the PBS-treated group among PD-1hiFoxp3−CD4+ T cells compared with PD-1−Foxp3−CD4+ T cells isolated from the tumors of 6C5-treated mice. f Tumor growth curves of B16-hAXL-bearing C57BL/6 mice (n = 6) treated with PBS (control), 6C5, or 6C5+anti-PD-1. g Tumor growth curves of B16-hAXL-bearing C57BL/6 mice treated with PBS (control, n = 5), 6C5 (n = 5), αPD-1 + αCTLA4 (n = 7), or 6C5 + αPD-1 + αCTLA4 (n = 8). h–k The frequency and number of intratumoral Tregs and PD-1hiFoxp3−CD4+ T cells were analyzed in B16-hAXL-bearing C57BL/6 mice treated with PBS (control), 6C5, αPD-1 + αCTLA4, or 6C5 + αPD-1 + αCTLA4. l Heatmap depicting the relative expression levels of the indicated gene sets among three intratumoral CD4⁺ T-cell subsets. m Tumor growth curves of B16-hAXL-bearing C57BL/6 mice treated with PBS (control, n = 5), 6C5 (n = 5), αPD-1-laIL2 (n = 7), or 6C5 + αPD-1-laIL2 (n = 6). n Tumor growth curves of C57BL/6 mice with complete tumor regression after treatment with 6C5 combined with αPD-1-laIL2, followed by subcutaneous rechallenge with B16-hAXL tumor cells. The data are presented as the means ± SEMs, and two or three independent experiments were performed. Statistical analysis of tumor growth was performed via two-way ANOVA. For (c, h–k), unpaired two-tailed t-tests were applied. ns (not significant), *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001
The combination of 6C5 with anti-PD-1 treatment did not result in a significantly enhanced therapeutic effect (Fig. 6f). We subsequently employed a combination therapy of 6C5 with dual immune checkpoint therapy (anti-PD-1 plus anti-CTLA4) in B16-hAXL melanoma (Fig. 6g). Notably, while dual ICIs only slightly inhibited tumor growth, the inclusion of 6C5 substantially improved therapeutic efficacy. Importantly, while individual treatment with 6C5 increased the intratumoral populations of both Tregs and PD-1hiFoxp3−CD4+ T cells (Fig. 6h–k), combination with dual ICIs selectively reduced the proportion and number of PD-1hiFoxp3−CD4+ T cells without significantly reducing the number of Tregs (Fig. 6h–k and Supplementary Fig. 5e). These findings highlighted that AXL Ab treatment synergized with dual ICIs and modulated distinct immunosuppressive CD4+ T cells for tumor control.
We further investigated the suppressive mechanisms of PD-1hiFoxp3−CD4+ T cells. Intratumoral CD4+ T cells were sorted for bulk RNA-seq and TCR sequencing. Transcriptomic profiling revealed that PD-1hiFoxp3−CD4+ T cells exhibit a distinct phenotype characterized by elevated expression of multiple immune checkpoint molecules, including CTLA4 (Ctla4), LAG3 (Lag3), PD-1 (Pdcd1), TIGIT (Tigit), and TIM-3 (Havcr2), as well as increased expression of immunosuppressive cytokines such as TGF-β (Tgfb1) and IL10 (Il10) (Fig. 6l). These results indicate that the PD-1hiFoxp3−CD4+ T subset exhibits a potent immunosuppressive phenotype. TCR sequencing further revealed that the majority of clonotypes within PD-1hiFoxp3−CD4+ T cells were unique to this subset, with minimal overlap with Tregs and even less overlap with PD-1−Foxp3−CD4+ T cells (Supplementary Fig. 5f). The top 10 clonotypes accounted for approximately 40% of the PD-1hiFoxp3−CD4+ T-cell repertoire (Supplementary Fig. 5g), but these dominant clonotypes were present at much lower frequencies or were undetectable in the Treg or PD-1−Foxp3−CD4+ T-cell subset (Supplementary Fig. 5h). Impressively, the top 10 clonotypes in PD-1−Foxp3−CD4+ T cells constituted approximately 80% of that subset (Supplementary Fig. 5g) and were scarcely detected in the other two subsets (Supplementary Fig. 5i). Together, these findings indicate that PD-1hiFoxp3−CD4+ T cells represent a clonally expanded and phenotypically distinct population, with only limited lineage overlap with tumor-infiltrating Tregs, and are largely unrelated to conventional PD-1−Foxp3−CD4+ T cells.
The proinflammatory cytokines produced by CD4+ T helper cells play crucial roles in the antitumor immune response.39 Among these cytokines, interleukin-2 (IL-2) is a pivotal cytokine that significantly influences the proliferation and activation of CD8+ T cells.40 To overcome the possible IL-2 insufficiency caused by the increase in intratumoral Tregs and PD-1hiFoxp3−CD4+ T cells induced by 6C5 treatment, we administered anti-PD-1-laIL2 to specifically deliver a low-affinity IL-2 variant into the TME.41 Notably, combination therapy with 6C5 and anti-PD-1-laIL2 resulted in significantly enhanced tumor control and complete tumor regression in approximately 50% of the treated mice (Fig. 6m).
CD4+ T-cell depletion has been previously shown to increase the efficacy of cancer immunotherapy but to compromise the development of antitumor immune memory.42 Similarly, in our study, it markedly amplified the antitumor activity of the membrane-proximal AXL-targeting antibody 6C5 (Fig. 3j) but failed to reject tumor rechallenge (Supplementary Fig. 5j), indicating a loss of durable immune protection. Impressively, all the mice that achieved complete tumor regression following treatment with 6C5 and anti-PD-1-laIL2 remained fully protected upon rechallenge (Fig. 6n). These findings suggest that combining AXL-targeted therapy with anti-PD-1-laIL2 therapy effectively counteracts immunosuppressive CD4+ T cells without depleting immune-supportive subsets, which would otherwise compromise immune memory, thereby achieving both robust tumor control and long-lasting immune protection.