
Cell culture
Human neural progenitor cells (hNPC) were prepared and cultured as described in Brnic et al. (2012)21, and differentiated into human neuronal and glial cells (hNGC) as described in Fares et al. (2020)20. Briefly, hNPC were seeded at a density of 44,000 cells/cm2 in culture plates coated with Matrigel™ (#354,230, Corning, USA). Differentiation into a mixed population of neuronal and glial cells was induced 24 h after plating by replacing N2A medium with 1:1 N2A and NBC media and withdrawing Epidermal Growth Factor (EGF, #PCYT-217, Eurobio Scientific, France) and basic Fibroblast Growth Factor (bFGF, #PCYT-218, Eurobio Scientific, France). N2A is composed of Advanced Dulbecco’s modified Eagle medium-F12 (#12,634,028, Gibco, Thermo Fisher Scientific, USA) supplemented with 2 mM L-glutamine (#25,030,081, Gibco, Thermo Fisher Scientific, USA), 0.1 mg/ml apo-transferrin (#T1147, Sigma-Aldrich, USA), 25 μg/ml insulin (#I9278, Sigma-Aldrich, USA), and 6.3 ng/ml progesterone (#P6149, Sigma-Aldrich, USA). NBC is composed of neurobasal medium (#21,103,049, Gibco, Thermo Fisher Scientific, USA) supplemented with 2 mM L-glutamine and B27 without vitamin A 1X (#12,587,010, Gibco, Thermo Fisher Scientific, USA). Differentiation conditions were maintained for 13 days with medium replacement twice a week, prior to infection. Ninety-six-well plates (#655,090, Greiner Bio-One, Austria) were used for fluorescent immunostaining, and 24-well plates (#353,047, Falcon, Corning, USA) were used to prepare lysates for RNA analysis.
Late cortical progenitor-like (LCP) cells were obtained from human induced pluripotent stem cells and differentiated into cortical glutamatergic neurons as described in Boissart et al. (2013)22. LCP were seeded at a density of 35,000 cells/cm2 in 384-well plates and differentiation conditions were maintained for 28 days.
VERO E6 (ATCC No. CRL-1586) cells were cultured in Dulbecco’s modified Eagle medium (#61,965,026, Gibco, Thermo Fisher Scientific, USA) supplemented with 10% fetal bovine serum (FBS, #CVFSVF00-01, Eurobio Scientific, France), 1% sodium pyruvate (#11,360,070, Gibco, Thermo Fisher Scientific, USA) and 1% penicillin–streptomycin (#15,140,122, Gibco, Thermo Fisher Scientific, USA).
Ethics approval and consent to participate
Human fetus was obtained after legal abortion with written informed consent from the patient. The procedure for the procurement and use of human fetal central nervous system tissue was approved and monitored by the “Comité Consultatif de Protection des Personnes dans la Recherche Biomédicale” of Henri Mondor Hospital, France. All methods were in compliance with relevant French laws and institutional guidelines. Authorization and declaration numbers from the French Research Ministry are AC-2017–2993 (CHU Angers) and DC-2019–3771 (UMR Virologie). The rabbit immunization protocol (anti-WNV-E3 antibody) complied with EU legislation (authorization 12/04/11–6 accorded by the ANSES/ENVA/UPEC ethical committee).
Virus and infection
Three different WNV strains were used: WNVNY99 (an American strain of lineage 1, Genbank Accession No. KC407666.1), WNVFR2015 (a European strain of lineage 1, Genbank Accession No. MT863559.1) and WNVFR2018 (a European strain of lineage 2, Genbank Accession No. MT863561.1). The three strains were kindly provided by Dr. Gaëlle Gonzalez (ANSES, Maisons-Alfort, France). Working stocks (Passage 4) were generated in VERO cells (VERO-ATCC-CCL81) cultured in DMEM medium, supplemented with 2% FBS. WNVFR2015 and WNVFR2018 were propagated once (at passage 2) in C6/36 cells. Titers were estimated by plaque assay on VERO cells as described in Donadieu et al. (2013)23.
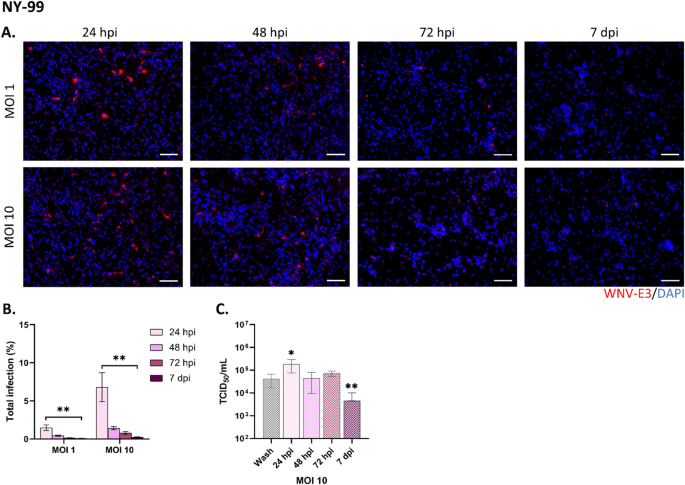
HNGCs differentiated for 13 days were infected at the indicated MOI or treated with culture medium only (“Mock”) for 90 min at 37 °C before removal of the inoculum. Subsequently, the cells were washed with 100 µL/well of fresh N2A/NBC medium. Immediately afterward, 60 µL/well was collected (called “wash”) and replaced by fresh medium until collection of supernatants and/or cell lysates at the indicated time points. Virus titers were estimated by endpoint dilution on VERO cells (TCID50), following the Reed and Muench method24. All procedures involving infectious materials were performed under bio-safety level-3 conditions.
Immunofluorescence assay
HNGC were fixed for 30 min in 4% paraformaldehyde (#15710, Electron Microscopy Sciences, USA) in PBS 1X and standard immunofluorescence was performed using antibodies for HuC/HuD (1:500, mouse, #A21271, Thermo Fisher Scientific, USA), βIII-tubulin (1:1000, mouse, #T8660, Sigma-Aldrich, USA or 1:1000, rabbit, #ab18207, Abcam, UK), Glial Fibrillary Acidic Protein (GFAP, 1:1000, mouse, #G3893, Sigma-Aldrich, USA or 1:1000, rabbit, Z0334, Dako, Denmark), Oligodendrocyte transcription factor 2 (OLIG2, 1:1000, goat, #AF2418, R&D Systems), cleaved caspase-3 (1:100, rabbit, #9661, Cell Signaling Technology, USA), S100β (1:1000, rabbit, #ab52642, Abcam, UK), double-stranded RNA (dsRNA, 1:800, mouse, #10020200, Scicons, Hungary) and the domain 3 of WNV envelope protein (WNV-E3, 1:1000, rabbit, in house). Cells were blocked for 2 h in 3% BSA (#A9647, Sigma-Aldrich, USA), 0.3% Triton-X-100 (VWR Chemical, Belgium) in PBS 1X. Primary antibodies were diluted in 0.3% BSA, 0.03% Triton-X-100 in PBS 1X, and incubated overnight at 4 °C. Secondary antibodies were Alexa Fluor-488/546/594-conjugated anti-mouse/anti-rabbit/anti-goat IgG (Molecular Probes, Invitrogen, Thermo Fisher Scientific, USA), diluted at 1:1000 and incubated for 2 h at room temperature. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (Life Technologies, Thermo Fisher Scientific, USA) at 0.1 ng/ml.
Image acquisition and analysis
The digitalized images shown were acquired with an AxioObserver Z1 (Zeiss, Germany) inverted microscope using ZEN software (v3.5, Zeiss, Germany) and were adjusted for brightness and contrast using this software.
To enumerate infected cells, three channel images were acquired in a fully automated and unbiased manner using the Opera Phenix™ Plus High-Content Screening System (Revvity, USA) and a 10 × air objective (NA = 0.3). Twelve images per channel per well (representing approximately 85% of the entire well) were acquired and analyzed with Signals Images Artist Analysis and Management software (SImA, Revvity, USA), using a customized algorithm for cell segmentation and identification. Briefly, nuclei were segmented based on DAPI staining. Living and dead cells were distinguished by the mean nuclear intensity, with dead cells exhibiting higher DAPI signal. Infected cells were enumerated by quantifying the intensity of WNV-E3 immunostaining in a perinuclear ring surrounding living nuclei. The threshold for WNV-E3 positivity was set at the lowest intensity value that reliably distinguished perinuclear WNV-E3 signal from background staining. Astrocytes were identified by the size of their nuclei (larger than those of neurons). Oligodendrocytes, were identified by immunostaining for OLIG2 in the nuclear region. Total infection refers to the percentage of astrocytes and oligodendrocytes infected relative to the total cell population. Astrocyte infection and oligodendrocyte infection refer to the percentage of infected cells within the astrocyte or oligodendrocyte populations, respectively.
For automated quantification of cells immunostained with antibodies directed against HuC/HuD and OLIG2 and of cell processes immunostained with antibodies against βIII-tubulin and GFAP, images were acquired using the ImageXpress micro automated microscope (Molecular Devices, UK) and analyzed using Custom Modules designed using MetaXpress Analysis Software V6).
Semi-quantitative quantification of cytokines in cell supernatant
The Proteome Profiler Human Cytokine Array kit (#ARY005B, R&D Systems, USA) was used to assess the impact of WNV infection on cytokine secretion in hNGC. It was used following the manufacturer’s instructions. Briefly, hNGC were cultured on 24-well plates and infected with WNVNY99 (MOI 10) for 24 h. Collected supernatants were pooled from three wells for each condition (500 µL/well) and were inactivated by UV-irradiation (254 nm, 2 J/cm2), using a CL-508 Crosslinker (Uvitec, UK). Inactivated supernatants (700 µL) were mixed with array buffers and the “Human Cytokine Array Detection Antibody Cocktail” before being incubated overnight at 4 °C with pre-blocked membranes spotted in duplicate with 36 antibodies for a variety of cytokines and chemokines (Supplementary table 1). Streptavidin-HRP was prepared at 1:2000 dilution in array buffer and added to the membranes for 30 min at room temperature. The array “Chemi Reagent Mix” was distributed evenly on each membrane before visualization with the ChemiDoc MP imaging system (Bio-Rad Laboratories, USA). Relative quantification was performed using ImageJ (v1.54 g) software by measuring the inverted grayscale intensity of each individual spot and normalizing it to the mean intensity of the designated reference spots.
Induction or inhibition of antiviral response in hNGC
To assess the impact of the IFN signaling pathway on WNV infection in hNGC, cells were pretreated with recombinant human IFN-β (100 U/mL, #11410–2, PBL Assay Science, USA) or with ruxolitinib (5 µM, #S1379, Selleck Chemicals LLC, USA), a JAK 1/2 inhibitor, for 2 h before infection with WNV (MOI 10). After removal of the inoculum, a fresh IFN-β or ruxolitinib dilution was added. At 24 h post-infection or treatment, cells were fixed or lysed and supernatants were harvested for subsequent analysis.
IFI6 downregulation
SiRNA targeting IFI6 was purchased from Horizon Discovery (Si-genome in SMARTpool format, #M-003672–02-0005). Human NGC cultured in 96-well plates were transfected with 25 nM of siRNA and 0.2 μL of DharmaFECT 1 Transfection reagent (Horizon Discovery, UK) as per manufacturer’s instructions. Forty-eight hours after transfection, RNAi-transfected cells were infected with WNVNY99 at an MOI of 10. Viral inoculum was removed 90 min later and replaced with 150 μL of fresh N2A/NBC medium. Cells were fixed and supernatants were collected 24 h post-infection. The impact of RNAi on viral infection was assessed by immunofluorescence labeling of infected cells and by quantification of WNV genomic RNA in supernatants by reverse transcriptase quantitative polymerase chain reaction (RT-qPCR).
RNA isolation and RT-qPCR
RNA was isolated from infected and non-infected hNGC. Cells were lysed and RNA extracted using the RNEasy mini kit (#74106, Qiagen, Germany), following the manufacturer’s instructions. Extraction of viral RNA from supernatants of infected cells was performed using QIAamp Viral RNA Mini Kit (#52904, Qiagen, Germany), according to the manufacturer’s instructions. One hundred nanograms of RNA from cell lysates and 2 μL of RNA from supernatant were used for cDNA synthesis using the SuperScript™ II Reverse Transcriptase kit (#18064022, Thermo Fisher Scientific, USA). Real-time PCR was performed in a total reaction volume of 10 µL, using 2 μL of cDNA and QuantiTect SYBR Green PCR master mix (Qiagen, Germany), on a LightCycler™ 96 instrument (Roche Applied Science, Germany). Samples were held for 15 min at 95 °C and then subjected to 40 amplification cycles consisting of incubations at 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 30 s. This was followed by a final step for melting curve analysis consisting of incubations at 95 °C for 10 s, 58 °C for 60 s, 96 °C for 1 s and 40 °C for 30 s. For relative quantification, the − 2ΔΔCt method was used25. GAPDH was used as the reference gene. Primers pairs are listed in Supplemental table 2.
Statistical analysis
Statistical analyses were performed using GraphPad Prism V10.0.0. Data normality was assessed using the Shapiro–Wilk test. Depending on the distribution and the experimental design, comparisons between two groups were performed using either an unpaired Student’s t test or a Mann–Whitney test. For comparisons between multiple time points, a one-way ANOVA analysis followed by a Tukey’s test or a Kruskal–Wallis test followed by a Dunn’s test was used. Statistical tests applied are specified in the legend of each figure.